
Mucopolysaccharidosis loại IV (mucopolysaccharidosis type IV) là bệnh di truyền xảy ra do enzyme lysosome giảm số lượng hoặc hoạt động bất thường dẫn đến tình trạng đường tích tụ trong cơ thể. Hiện tượng này làm tổn thương tế bào, mô và nhiều cơ quan trong cơ thể, gây các biểu hiện lâm sàng liên quan đến xương, hệ hô hấp và tim mạch.
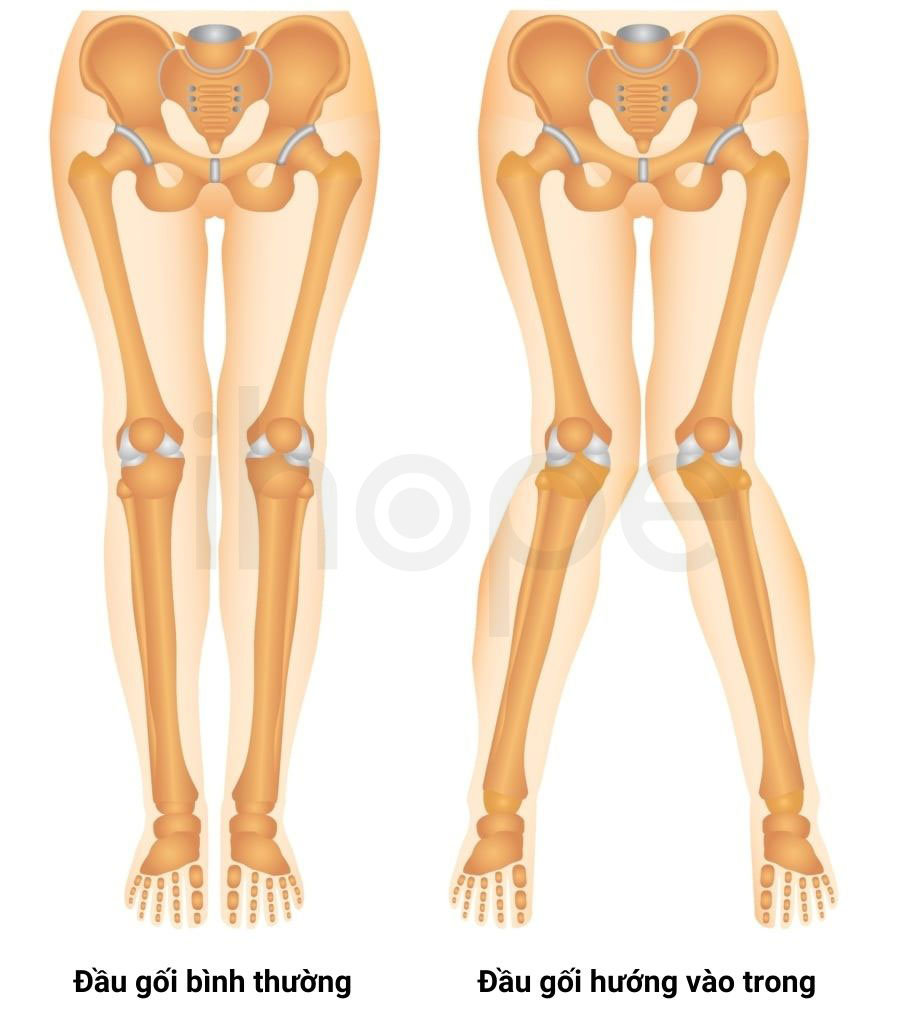
Ảnh: Hai đầu gối hướng vào trong
Nguồn: MyMSK Clinic
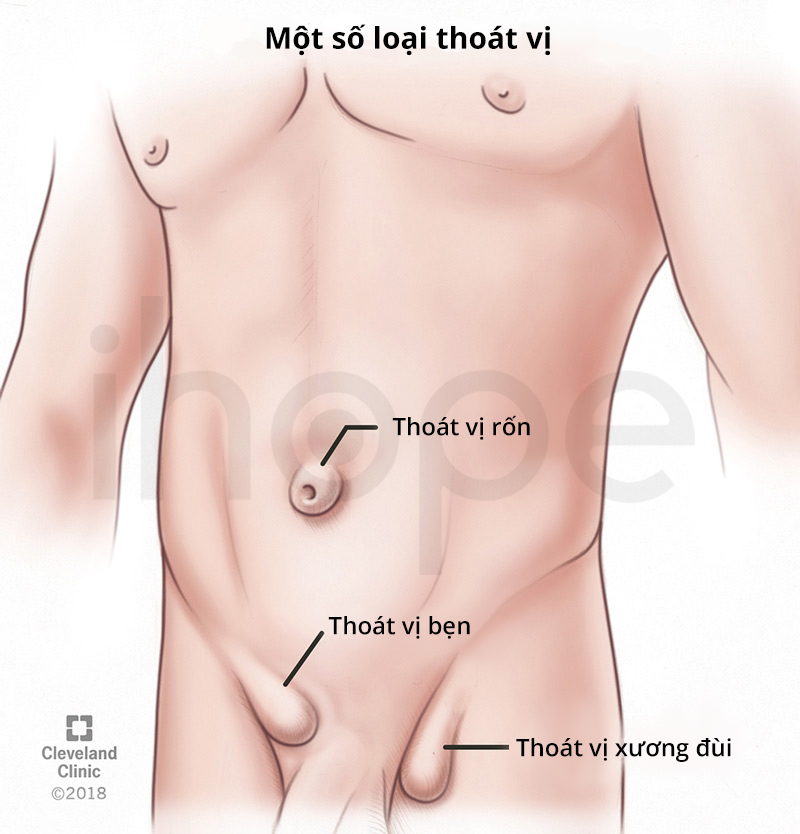
Ảnh: Một số loại thoát vị
Nguồn: Cleveland Clinic
Biểu hiện lâm sàng
Người mắc hội chứng mucopolysaccharidosis loại IV thường có các triệu chứng sau:
Bất thường xương
- Tầm vóc thấp
- Đầu gối hướng vào trong
- Dị dạng xương sườn, ngực, cột sống, hông, cổ tay
- Khớp lỏng lẻo và linh hoạt quá mức hoặc vận động kém
- Mỏm nha kém phát triển
Trong đó, mỏm nha (cấu trúc nhô lên từ thân của đốt sống cổ thứ hai) kém phát triển có thể dẫn đến sai khớp đốt sống cổ, từ đó gây chèn ép và tổn thương tủy sống. Một số trường hợp nghiêm trọng có thể dẫn đến liệt hoặc thậm chí tử vong.
Triệu chứng khác
Người mắc bệnh mucopolysaccharidosis loại IV có thể biểu hiện một số triệu chứng khác như:
- Đục giác mạc dẫn đến giảm thị lực
- Nhiễm trùng tai, giảm thính lực
- Hẹp đường thở gây nhiễm trùng đường hô hấp trên và ngưng thở khi ngủ
- Khuôn mặt dị biệt
- Mòn men răng, sâu răng
- Dị tật van tim
- Gan lớn
- Thoát vị rốn hoặc bẹn
Độ phổ biến
Ước tính tỉ lệ mắc hội chứng mucopolysaccharidosis loại IV trên toàn thế giới khoảng 1/200.000–300.000 người.
Nguyên nhân
Mucopolysaccharidosis loại IV được chia thành hai dạng dựa trên gen đột biến gây bệnh, bao gồm GALNS gây mucopolysaccharidosis loại IVA và GLB1 gây mucopolysaccharidosis loại IVB. Những gen này cung cấp hướng dẫn tạo ra các enzyme tham gia vào quá trình phân giải những phân tử đường lớn glycosaminoglycan (GAG). Hai dạng của hội chứng đều có các biểu hiện lâm sàng giống nhau nên khó phân biệt nếu chỉ dựa trên triệu chứng bệnh.
Đột biến một trong hai gen này có thể làm giảm hoặc mất hoàn toàn hoạt tính của enzyme nên GAG ngừng phân giải, thay vào đó chúng tích tụ trong tế bào đặc biệt là bên trong lysosome—bào quan giữ vai trò phân giải và tái chế các phân tử như carbohydrate hay lipid. Khi GAG tích tụ đến ngưỡng nhất định, chúng có thể gây hại cho nhiều mô và cơ quan như xương, từ đó một số đặc điểm dị dạng xương trong hội chứng mucopolysaccharidosis loại IV xuất hiện. Ngoài ra, hiện tượng tích tụ GAG còn cản trở hoạt động của những protein khác trong lysosome cũng như ảnh hưởng quá trình vận chuyển các phân tử trong tế bào để góp phần gây ra những biểu hiện lâm sàng khác của hội chứng.
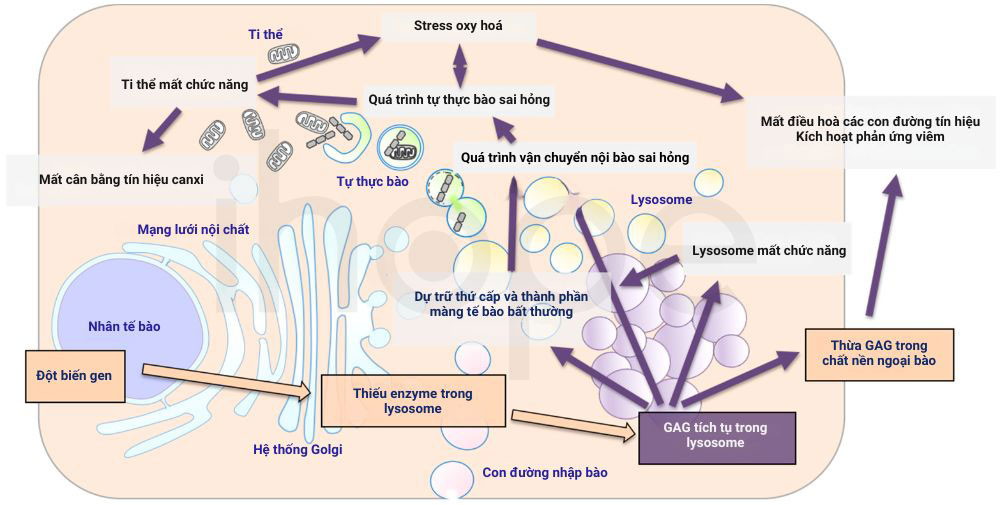
Nguồn: MDPI
Chẩn đoán
Mucopolysaccharidosis loại IV được chẩn đoán thông qua một số phương pháp sau:
- Đánh giá biểu hiện lâm sàng, bệnh sử: quan sát những triệu chứng liên quan đến phát triển thần kinh, vận động và các đặc điểm bất thường quanh vùng đầu
- Chụp X-quang: phát hiện dị tật xương đặc trưng của hội chứng
- Xét nghiệm nước tiểu: kiểm tra nồng độ glycosaminoglycan
- Xét nghiệm di truyền: xác định đột biến gen GALNS và GLB1 nhằm xác nhận chẩn đoán.
Điều trị
Hiện nay, chưa có phương pháp điều trị hoàn toàn hội chứng mucopolysaccharidosis loại IV. Các liệu pháp chủ yếu nhằm giảm nhẹ triệu chứng và cải thiện chất lượng đời sống người bệnh như:
- Sử dụng xe lăn hoặc nạng nhằm hỗ trợ di chuyển
- Phẫu thuật chỉnh hình nhằm cố định đốt sống cổ
- Trị liệu phục hồi khả năng vận động, hỗ trợ tinh thần
- Tái tạo mạch khí quản đối với trường hợp tắc nghẽn khí quản nguy kịch
Dạng di truyền
Hội chứng mucopolysaccharidosis loại IV di truyền theo kiểu trội trên nhiễm sắc thể thường. Do đó, một bản sao của gen đột biến trong mỗi tế bào đủ để gây ra bệnh. Người bệnh thường được di truyền từ cha hoặc mẹ mắc bệnh. Một số ít trường hợp là do đột biến mới (de novo) xuất hiện trong quá trình hình thành tế bào sinh sản (trứng, tinh trùng) hoặc quá trình phát triển phôi sớm.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Hội chứng mucopolysaccharidosis loại IV di truyền trội trên nhiễm sắc thể thường, nếu cha hoặc mẹ mắc bệnh, con sinh ra sẽ có 50% khả năng di truyền bệnh. Do đó, để bảo đảm 100% khả năng con không bị bệnh, cha mẹ có thể chọn phương pháp thụ tinh nhân tạo IVF và sàng lọc phôi PGS/PGD. Các thành viên trong gia đình nên khám sức khỏe và tầm soát bệnh định kỳ nếu có thành viên mắc bệnh. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh.
Các tên gọi khác
- Morquio disease
- Morquio syndrome
- Morquio's disease
- Morquio's syndrome
- Morquio-Brailsford disease
- MPS IV Mucopolysaccharidosis (MPS) IV (A, B)
References
- National Library of Medicine. Morquio syndrome. Retrieved September 10, 2025 from https://www.ncbi.nlm.nih.gov/books/NBK564330/
- Genetic and Rare Diseases Information Center. Mucopolysaccharidosis type 4a. Retrieved September 10, 2025 from https://rarediseases.info.nih.gov/diseases/3785/index
- Catalog of Genes and Diseases from OMIM. MUCOPOLYSACCHARIDOSIS, TYPE IVA; MPS4A. Retrieved September 10, 2025 from https://omim.org/entry/253000
- U.S National Library of Medicine. Mucopolysaccharidosis type IV. Retrieved September 10, 2025 from https://medlineplus.gov/genetics/condition/mucopolysaccharidosis-type-iv/
- National Organisation for Rare Disorders. Mucopolysaccharidosis IV. Retrieved September 10, 2025 from https://rarediseases.org/rare-diseases/morquio-syndrome/
- Orphanet. Mucopolysaccharidosis type 4. Retrieved September 10, 2025 from https://www.orpha.net/en/disease/detail/582





