Mucopolysaccharidosis loại VII (mucopolysaccharidosis type VII) hay hội chứng Sly là bệnh di truyền tác động chủ yếu đến mô và cơ quan. Mức độ nghiêm trọng của bệnh khác nhau giữa từng người.
Biểu hiện lâm sàng
Dấu hiệu nghiêm trọng nhất của bệnh mucopolysaccharidosis loại VII là phù thai—hiện tượng cơ thể tích tụ chất lỏng dư thừa trước khi sinh. Đa số trẻ sơ sinh phù thai đều chết lưu hoặc tử vong ngay sau khi chào đời. Đối với các trường hợp sống sót, bệnh thường khởi phát trong giai đoạn trẻ nhỏ.
Biểu hiện lâm sàng của bệnh bao gồm:
- Đầu to
- Não úng thủy
- Khuôn mặt thô
- Lưỡi to
- Gan và lách to
- Triệu chứng liên quan đến van tim
- Thoát vị rốn
- Thoát vị bẹn
- Nhiễm trùng tai tái phát
- Mất thính lực
- Chậm phát triển
- Thiểu năng trí tuệ tiến triển
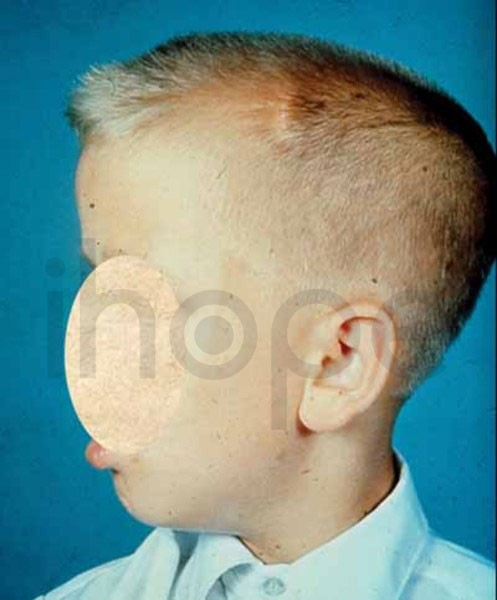
Ảnh: Dị tật đầu to
Nguồn: National Library of Medicine
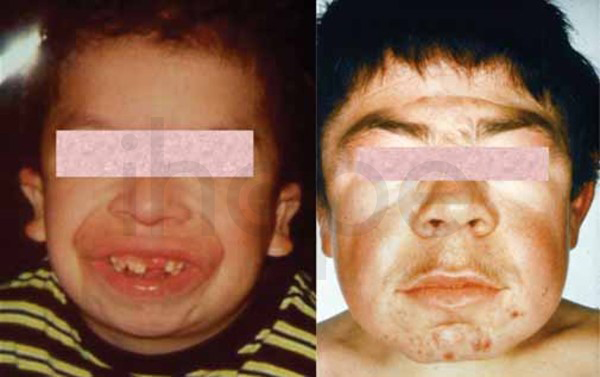
Ảnh: Đặc điểm khuôn mặt dị biệt
Nguồn: Elements of Morphology, National Human Genome Research Institute
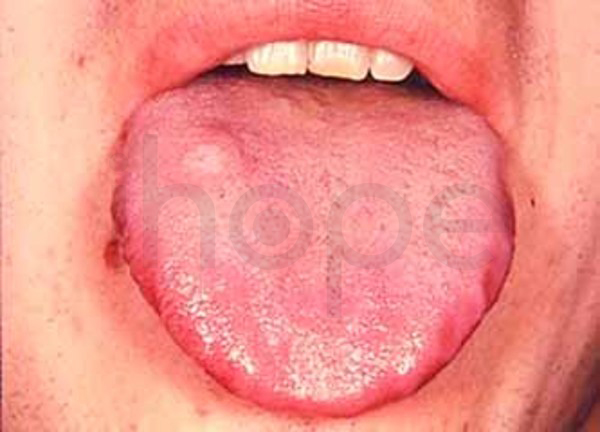
Ảnh: Lưỡi lớn
Nguồn: Elements of Morphology, National Human Genome Research Institute
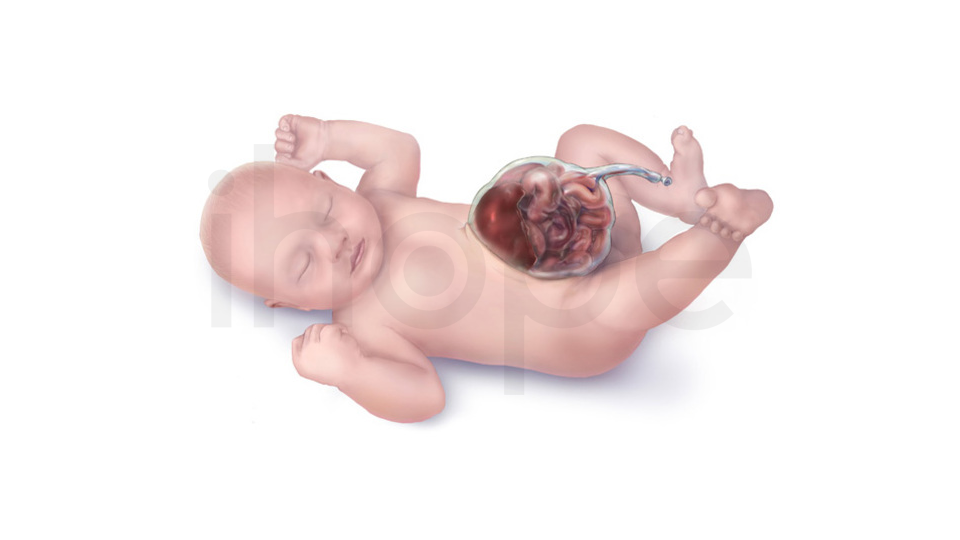
Ảnh: Thoát vị rốn
Nguồn: Shutterstock
Ngoài ra, bệnh nhân còn có đường thở hẹp hơn bình thường dẫn đến dễ nhiễm trùng đường hô hấp trên và ngừng thở thời gian ngắn trong lúc ngủ. Bên cạnh đó, người bệnh có nguy cơ mất thị lực do giác mạc đục dần.
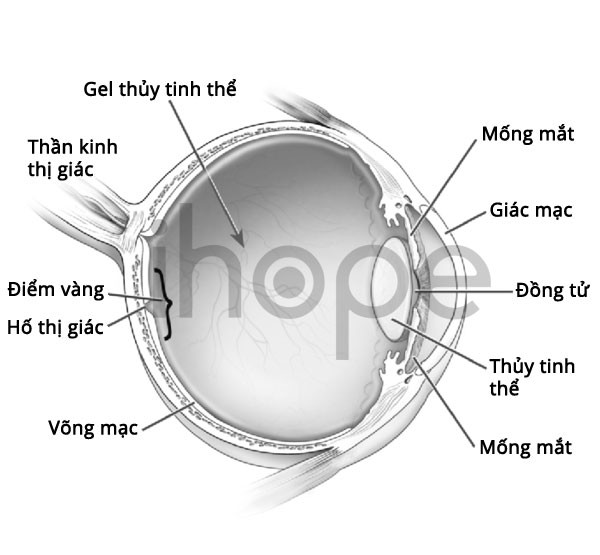
Nguồn: National Institutes of Health
Mặc khác, bệnh khiến xương dị dạng và tiến triển nghiêm trọng theo thời gian. Các biểu hiện về xương bao gồm vóc dáng nhỏ và biến dạng khớp làm ảnh hưởng đến khả năng vận động. Người bệnh còn có dấu hiệu loạn sản xương đa ổ do quá trình phát triển không ổn định của xương. Trong một số trường hợp, trẻ bệnh xuất hiện hội chứng ống cổ tay.
Dấu hiệu của hội chứng ống cổ tay gồm có:
- Tê tay
- Ngứa ran
- Bàn tay và ngón tay yếu
Ngoài ra, người bệnh có nguy cơ hẹp ống sống cổ gây chèn ép và tổn thương tủy sống. Tuổi thọ của người bệnh phụ thuộc vào mức độ nghiêm trọng của các triệu chứng. Một số bệnh nhân không sống qua giai đoạn trẻ nhỏ, trong khi những người khác có thể sống đến tuổi vị thành niên hoặc trưởng thành. Bệnh tim và tắc nghẽn đường thở là nguyên nhân chính gây tử vong cho người bệnh.
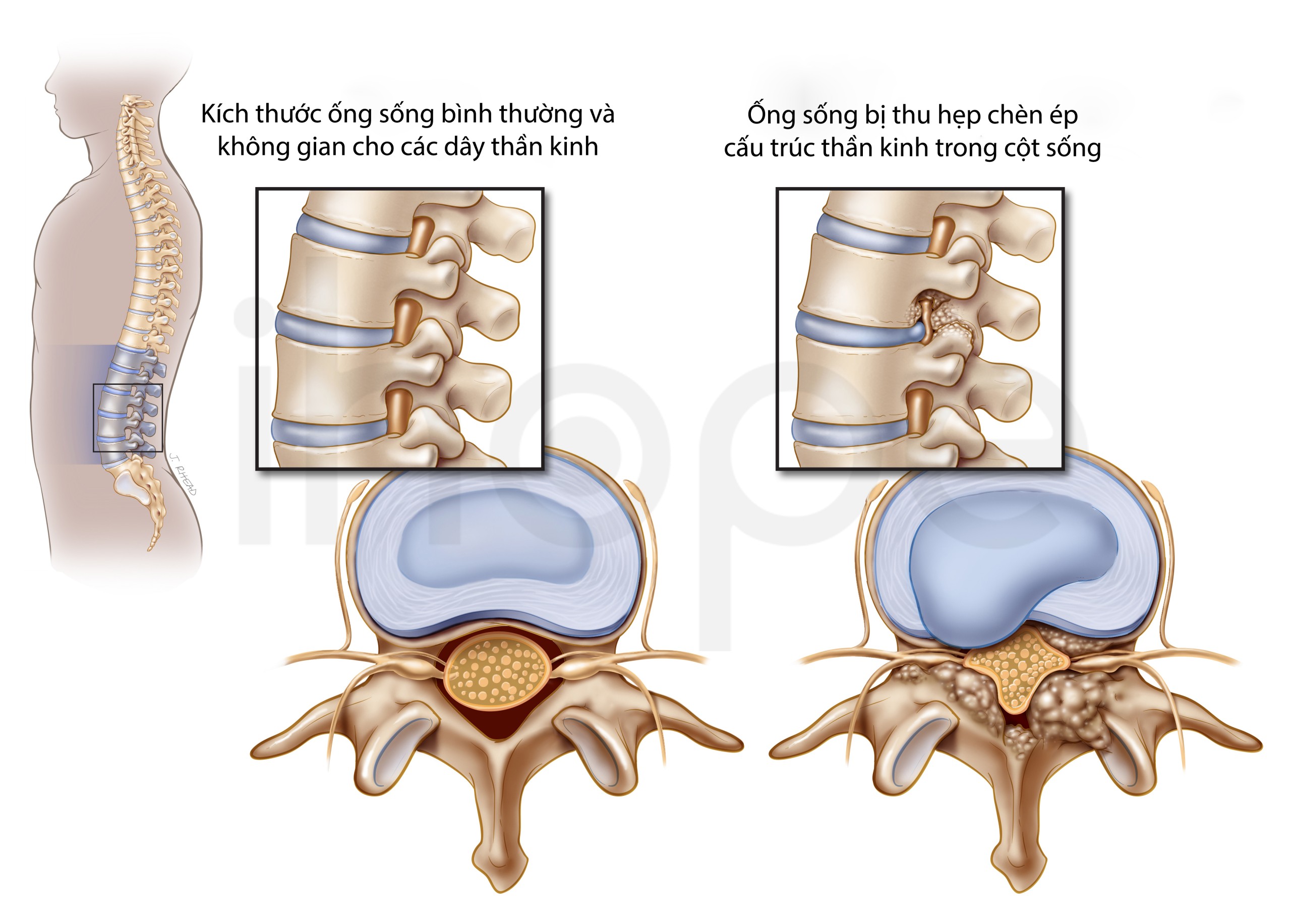
Nguồn: intermountain healthcare.org
Độ phổ biến
Hiện nay, tỉ lệ mắc bệnh mucopolysaccharidosis loại VII vào khoảng 1/250.000 trẻ sơ sinh. Đây là loại hiếm gặp nhất của nhóm bệnh mucopolysaccharidosis.
Nguyên nhân
Đột biến gen GUSB gây ra bệnh mucopolysaccharidosis loại VII. Gen GUSB cung cấp hướng dẫn tạo ra enzyme beta-glucuronidase có chức năng phân giải các phân tử đường lớn như glycosaminoglycan (GAG). Glycosaminoglycan là các mucopolysaccharide.
Đột biến gen GUSB khiến enzyme beta-glucuronidase giảm hoặc không hoạt động. Thiếu enzyme beta-glucuronidase dẫn đến tích tụ glycosaminoglycan trong tế bào, chủ yếu trong lysosome. Lysosome là bào quan có chức năng phân giải và tái sử dụng nhiều loại phân tử. Quá trình tích tụ các phân tử trong lysosome gọi là bệnh tích trữ lysosome. Lượng glycosaminoglycan tích tụ làm tăng kích thước của lysosome khiến mô và cơ quan trở nên phình đại. Đồng thời, glycosaminoglycan cũng ảnh hưởng đến chức năng của protein khác trong lysosome và làm gián đoạn nhiều hoạt động của tế bào.
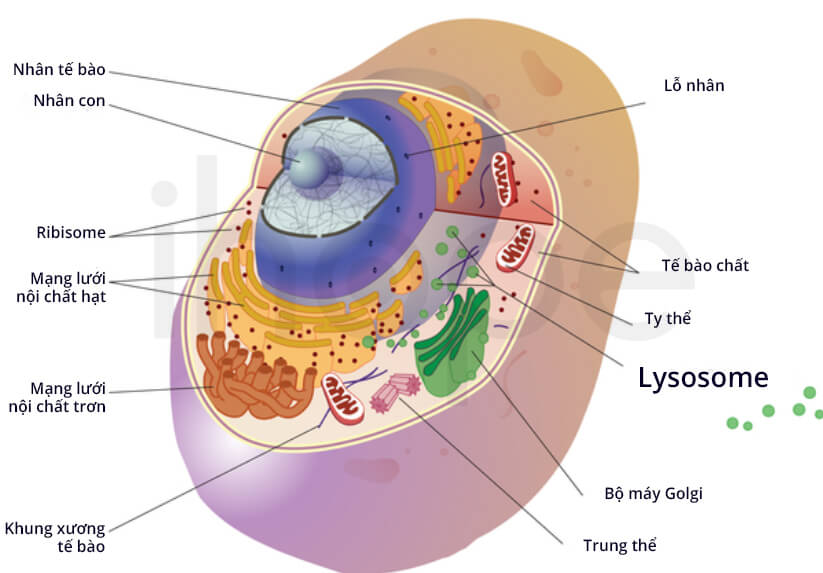
Nguồn: Darryl Leja, NHGRI
Chẩn đoán
Bệnh mucopolysaccharidosis loại VII được chẩn đoán dựa trên triệu chứng lâm sàng và bệnh sử gia đình. Ngoài ra, bác sĩ cũng chỉ định thực hiện xét nghiệm sinh hóa nhằm kiểm tra các chỉ số liên quan đến nồng độ mucopolysaccharide trong nước tiểu. Đa số bệnh nhân có nồng độ dermatan sulfate, heparan sulfate và chondroitin sulfate tăng cao.
Ngoài ra, người bệnh có thể thực hiện xét nghiệm chuyên khoa để đo mức độ hoạt động của beta-glucuronidase trong máu hoặc tế bào da.
Đối với thai phụ, bác sĩ thường thực hiện chẩn đoán trước sinh bằng phương pháp chọc ối hoặc lấy mẫu nhung mao màng đệm. Trong trường hợp cần thiết, bác sĩ sẽ chỉ định bệnh nhân xét nghiệm di truyền để xác nhận chẩn đoán.
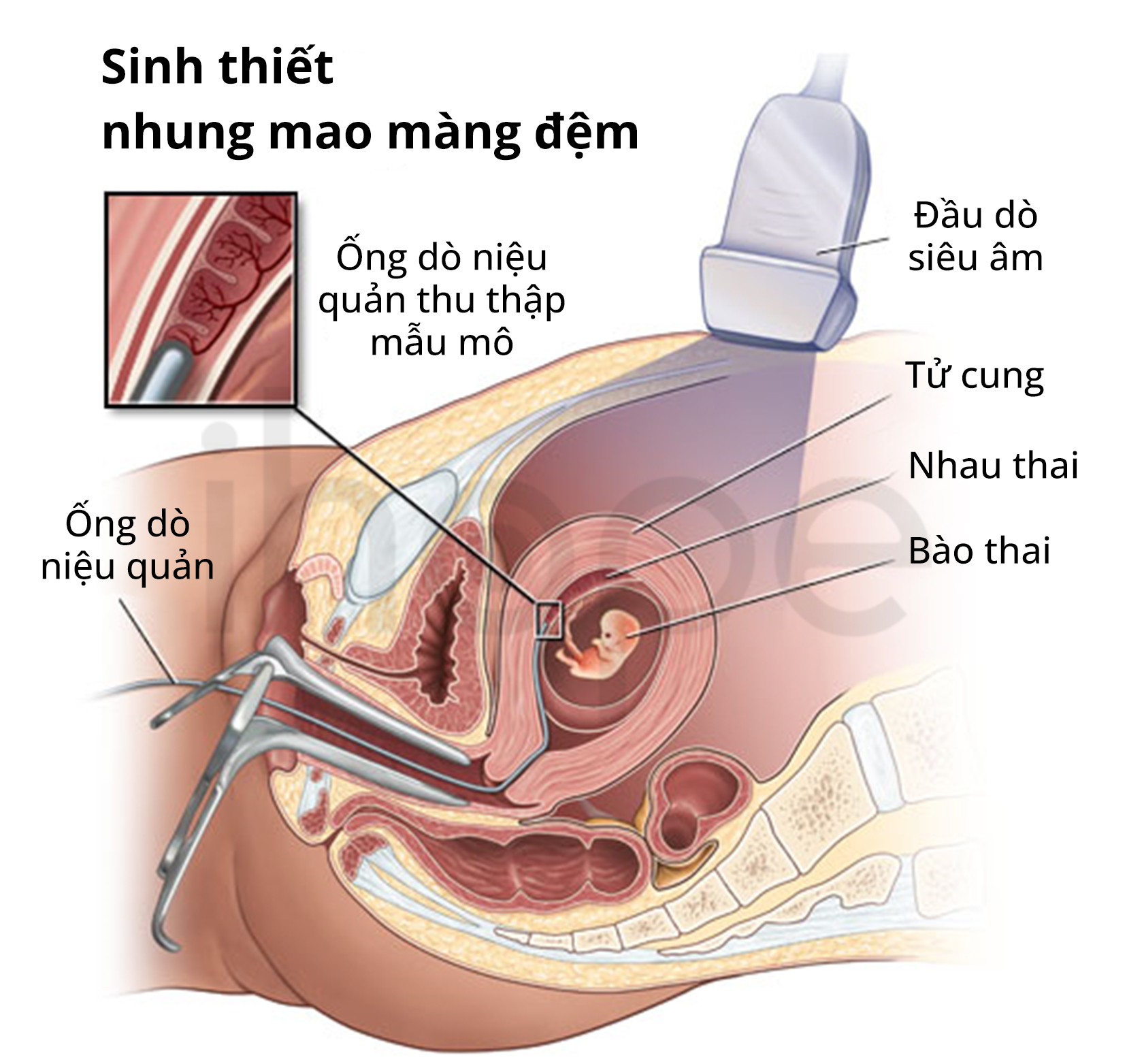
Nguồn: Johns Hopkins Medicine
Điều trị
Hiện nay, chưa có liệu pháp điều trị hoàn toàn bệnh mucopolysaccharidosis loại VII. Các liệu pháp chủ yếu làm giảm triệu chứng bệnh và cải thiện cuộc sống người bệnh.
Bệnh nhân cần thực hiện phẫu thuật trong các trường hợp sau:
- Xương biến dạng
- Thoát vị
- Dị tật mắt và tim
Ngoài ra, người dị tật đường thở hoặc cột sống cổ cũng cần phẫu thuật. Tuy nhiên, bệnh nhân cần kiểm tra độ nhạy cảm với thuốc mê và các biện pháp phòng ngừa trước khi thực hiện phẫu thuật.
Hiện nay, liệu pháp sử dụng enzyme thay thế hay liệu pháp Mepsevii đã được công nhận. Ngoài ra, một liệu pháp khác cũng mang lại nhiều kết quả tích cực là liệu pháp tế bào gốc tạo máu. Những người áp dụng liệu pháp này có mức độ tiến triển bệnh chậm hoặc gần như không có các biến chứng thần kinh. Phần lớn bệnh nhân sử dụng liệu pháp này từ giai đoạn đầu thường không xuất hiện dấu hiệu về xương.
Dạng di truyền
Bệnh mucopolysaccharidosis loại VII di truyền theo kiểu lặn trên nhiễm sắc thể thường, nghĩa là cả hai bản sao của gen trong mỗi tế bào đều có đột biến. Cha mẹ của một cá nhân mắc bệnh lặn trên nhiễm sắc thể thường đều mang một bản sao của gen đột biến, nhưng họ thường không biểu hiện dấu hiệu và triệu chứng của bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Bệnh mucopolysaccharidosis loại VII di truyền lặn đột biến gen GUSB, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kì.
Các tên gọi khác
- Beta-glucuronidase deficiency
- GUSB deficiency
- MPS VII
- MPS7
- Mucopolysaccharidosis 7
- Mucopolysaccharidosis VII
- Sly Syndrome
References
- Genetic and Rare Diseases Information Center. Mucopolysaccharidosis type 7. Retrieved September 06, 2024 from https://rarediseases.info.nih.gov/diseases/7096/index
- Catalog of Genes and Diseases from OMIM. MUCOPOLYSACCHARIDOSIS, TYPE VII; MPS7. Retrieved September 06, 2024 from https://omim.org/entry/253220
- U.S. National Library of Medicine. Mucopolysaccharidosis type VII. Retrieved September 06, 2024 from https://medlineplus.gov/genetics/condition/mucopolysaccharidosis-type-vii/
- Genetic Testing Information. Mucopolysaccharidosis type 7. Retrieved September 06, 2024 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0085132/
- National Institute of Health. Clinical course of sly syndrome (mucopolysaccharidosis type VII). Retrieved September 06, 2024 from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4893087/
- National Organization for Rare Disorders. Mucopolysaccharidosis Type VII. Retrieved September 06, 2024 from https://rarediseases.org/rare-diseases/sly-syndrome/
- Orphanet. Mucopolysaccharidosis type 7. Retrieved September 06, 2024 from https://www.orpha.net/en/disease/detail/584
- Dove Medical Press. Diagnosis and Emerging Treatment Strategies for Mucopolysaccharidosis VII (Sly Syndrome). Retrieved September 06, 2024 from https://www.dovepress.com/diagnosis-and-emerging-treatment-strategies-for-mucopolysaccharidosis--peer-reviewed-fulltext-article-TCRM
- Boston Children's Hospital. Mucopolysarcharidosis Type VII (MPS VII). Retrieved September 06, 2024 from https://www.childrenshospital.org/conditions/mucopolysarcharidosis-type-vii