
Rối loạn glycosyl hóa bẩm sinh COG5 (COG5-congenital disorder of glycosylation) là bệnh di truyền hiếm gặp, người bệnh gặp các vấn đề thần kinh và nhiều bất thường khác. Mức độ nghiêm trọng các triệu chứng khác nhau giữa từng bệnh nhân.
Biểu hiện lâm sàng
Bệnh thường khởi phát các triệu chứng từ giai đoạn trẻ nhỏ. Người bệnh bị yếu cơ và chậm phát triển dẫn đến tầm vóc thấp. Những vấn đề thần kinh bao gồm thiểu năng trí tuệ trung bình đến nặng, khả năng phối hợp vận động kém và đi lại khó khăn. Một số trường hợp mắc bệnh mất khả năng nói.
Ngoài ra, bệnh nhân có gương mặt dị biệt bao gồm:
- Dị tật đầu nhỏ
- Tai thấp, xoay về sau
- Cổ ngắn
- Đường chân tóc phía sau gáy thấp
- Mũi to
Ảnh: Dị tật đầu nhỏ
Nguồn: Elements of Morphology, National Human Genome Research Institute
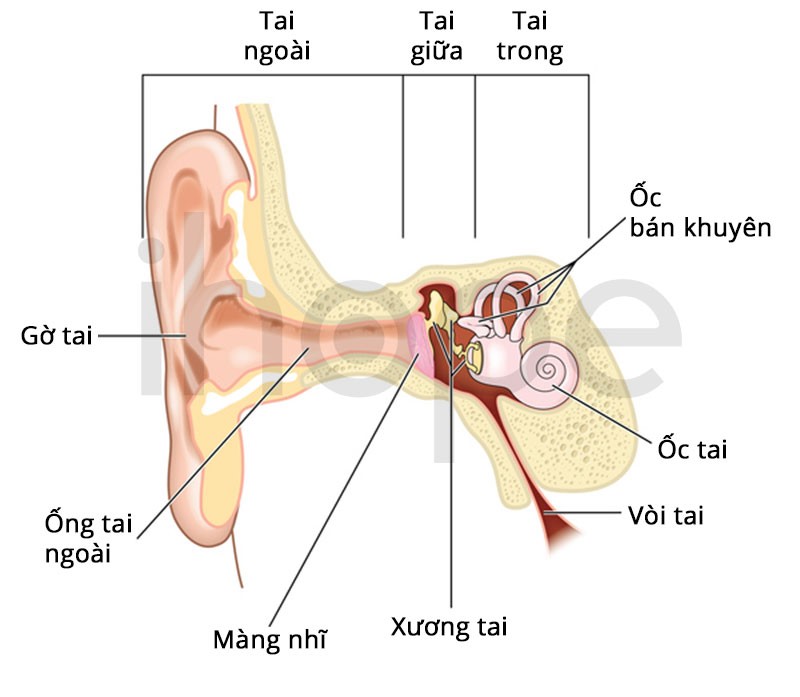
Hiếm hơn, người bệnh phát triển chứng mất thính giác do bất thường cấu trúc tai trong , suy giảm thị lực, tổn thương dây thần kinh kiểm soát chức năng bàng quang (bàng quang thần kinh), bệnh gan và biến dạng khớp.
Ảnh: Dị tật đầu nhỏ
Nguồn: Shutterstock
Độ phổ biến
Rối loạn glycosyl hóa COG5 bẩm sinh hiếm gặp, do đó hiện nay tỷ lệ mắc bệnh vẫn chưa được thống kê cụ thể. Khoảng 10 trường hợp mắc bệnh được báo cáo trong các tài liệu y khoa.
Nguyên nhân
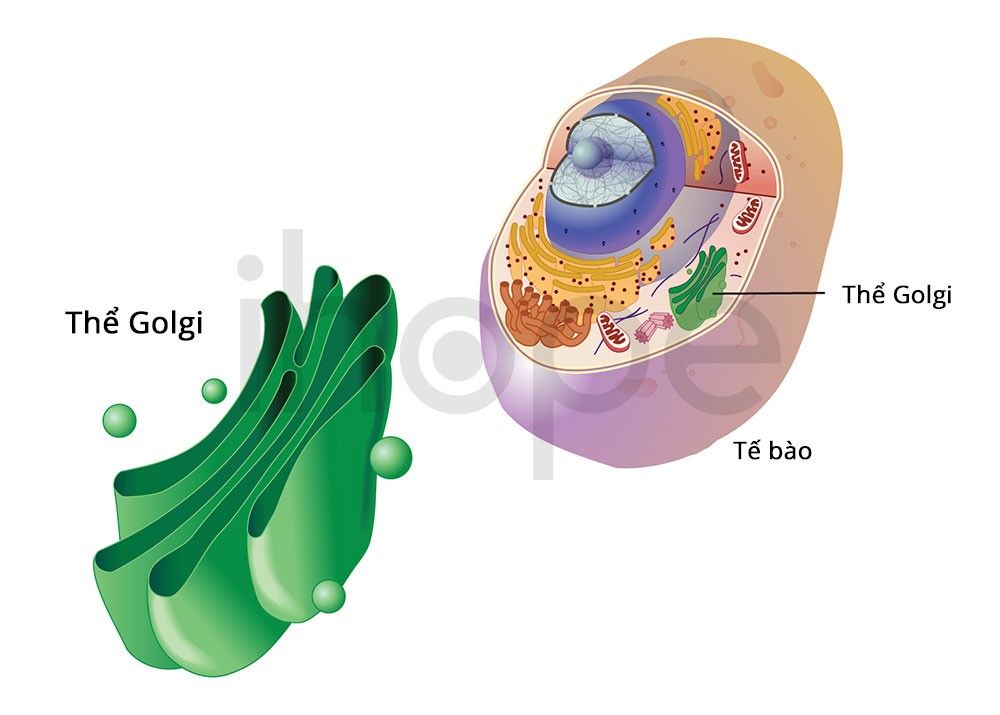
Đột biến gen COG5 gây ra rối loạn glycosyl hóa COG5 bẩm sinh. Gen COG5 cung cấp hướng dẫn tạo ra một tiểu đơn vị thuộc phức hợp oligomeric Golgi (COG) hoạt động trong bộ máy Golgi. Quá trình glycosyl hóa xảy ra trong bộ máy Golgi, các phân tử protein và chất béo gắn thêm các phân tử đường (oligosaccharide), từ đó protein thực hiện được nhiều chức năng hơn.
Ảnh: National Human Genome Research Institute
Phức hợp COG tham gia vận chuyển protein, kể cả những protein thực hiện quá trình glycosyl hóa trong bộ máy Golgi. Đột biến gen COG5 giảm hoặc mất hoàn toàn lượng protein COG5 dẫn đến gián đoạn vận chuyển protein. Do đó, quá trình glycosyl hóa bất thường ảnh hưởng nhiều cơ quan, dẫn đến các dấu hiệu và triệu chứng của bệnh. Mức độ nghiêm trọng của bệnh phụ thuộc vào lượng protein COG5 hiện diện trong tế bào.
Chẩn đoán
Rối loạn glycosyl hóa bẩm sinh COG5 hiếm gặp, do đó công tác chẩn đoán gặp nhiều khó khăn. Bác sĩ chẩn đoán dựa vào biểu hiện lâm sàng bao gồm chậm phát triển, tầm vóc thấp bé, tật đầu nhỏ, thiểu năng trí tuệ và khuôn mặt dị biệt. Xét nghiệm di truyền tìm đột biến gen COG5 thông qua mẫu máu hoặc mẫu phết niêm mạc miệng.
Điều trị
Hiện tại, chưa có phương pháp điều trị hoàn toàn rối loạn glycosyl hóa bẩm sinh COG5. Mục tiêu điều trị chủ yếu giảm thiểu các triệu chứng bệnh, từ đó cải thiện chất lượng cuộc sống của người bệnh.
Các phương pháp điều trị hiện tại bao gồm:
- Điều trị các triệu chứng về tiêu hóa, rối loạn tiểu tiện, rối loạn thần kinh
- Tập vật lý trị liệu để cải thiện khả năng vận động, tăng sức mạnh cơ bắp, tăng cường cân bằng và giảm thiểu các vấn đề về khớp
- Chế độ dinh dưỡng lành mạnh, đầy đủ các nhóm chất
Bên cạnh đó, người bệnh cần theo dõi và tầm soát biến chứng, ví dụ như đường huyết, chức năng gan và vấn đề hô hấp.
Dạng di truyền
Rối loạn glycosyl hóa bẩm sinh COG5 di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh lặn trên nhiễm sắc thể thường sẽ có bố và mẹ mang một bản sao của gen đột biến, nhưng bố mẹ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Bệnh di truyền lặn đột biến gen COG5, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kỳ.
Các tên gọi khác
- Carbohydrate deficient glycoprotein syndrome type IIi
- CDG IIi
- CDG2I
- CDGIIi
- COG5-CDG
- Congenital disorder of glycosylation type IIi
- CDG syndrome type IIi
- Congenital disorder of glycosylation type 2i
References
- Genetic Testing Information. COG5-congenital disorder of glycosylation. Retrieved March 23, 2023 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C3150876/
- Genetic and Rare Diseases Information Center. COG5-CDG (CDG-IIi). Retrieved March 23, 2023 from https://rarediseases.info.nih.gov/diseases/12348/cog5-cdg-cdg-iii
- Catalog of Genes and Diseases from OMIM. CONGENITAL DISORDER OF GLYCOSYLATION, TYPE Iii. Retrieved March 23, 2023 from https://omim.org/entry/613612
- U.S National Library of Medicine. COG5-congenital disorder of glycosylation. Retrieved March 23, 2023 from https://medlineplus.gov/genetics/condition/cog5-congenital-disorder-of-glycosylation/
- MalaCards. Cog5-Congenital Disorder of Glycosylation. Retrieved March 23, 2023 from https://www.malacards.org/card/cog5_congenital_disorder_of_glycosylation
- National Institute of Health. Identification of Two Novel Mutations in COG5 Causing Congenital Disorder of Glycosylation. Retrieved March 23, 2023 from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7056739/
- National Organization for Rare Disorders. COG5-CDG (CDG-IIi). Retrieved March 23, 2023 from https://rarediseases.org/gard-rare-disease/cog5-cdg-cdg-iii/
- Orphanet. COG5-CDG. Retrieved March 23, 2023 from https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=263487
- Fung CW, Matthijs G, Sturiale L, Garozzo D, Wong KY, Wong R, Wong V, Jaeken J. COG5-CDG with a Mild Neurohepatic Presentation. JIMD Rep. 2012;3:67-70. doi: 10.1007/8904_2011_61





