
Hội chứng Trichorhinophalangeal loại I (Trichorhinophalangeal syndrome type I) là bệnh gây dị dạng xương và khớp. Tên của bệnh mô tả một số vùng trên cơ thể bị ảnh hưởng, bao gồm tóc (tricho-), mũi (rhino-), ngón tay và ngón chân (phalangeal). Người bệnh có các biểu hiện đặc trưng bao gồm khuôn mặt dị biệt, thiểu năng trí tuệ và các bất thường khác.
Biểu hiện lâm sàng
Đầu xương ngón tay hoặc ngón chân của người bệnh có hình nón bất thường. Ngoài ra, móng tay và móng chân mỏng và biến dạng. Người bệnh có bàn chân ngắn .
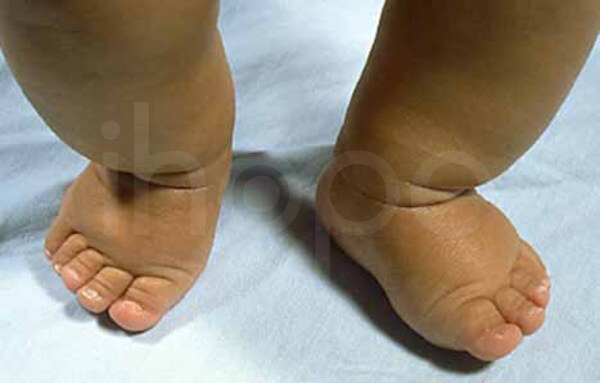
Ảnh: Bàn chân ngắn
Nguồn: National Human Genome Research Institute
Trẻ mắc bệnh có phạm vi cử động các khớp lớn hơn trẻ khỏe mạnh. Theo thời gian, các khớp bị thoái hóa làm hạn chế cử động và đau khớp. Lệch khớp háng (loạn sản xương hông) thường thấy vào giai đoạn tuổi trưởng thành.

Ảnh: Khớp ngón tay linh động
Nguồn: Mitskevich Uladzimir/Shutterstock.com
Các đặc điểm trên khuôn mặt, bao gồm:
- Lông mày rậm
- Mũi rộng, đầu tròn
- Tai lớn
- Nhân trung nhân rộng và phẳng
- Môi trên mỏng
- Răng nhỏ và nhiều
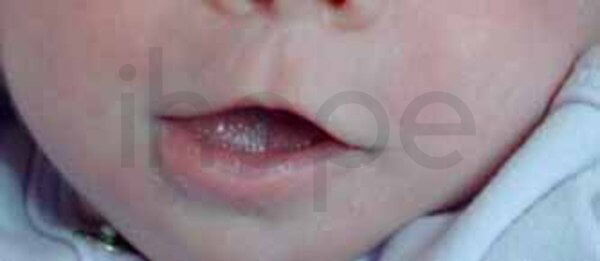
Ảnh: Môi trên mỏng
Nguồn: National Human Genome Research Institute
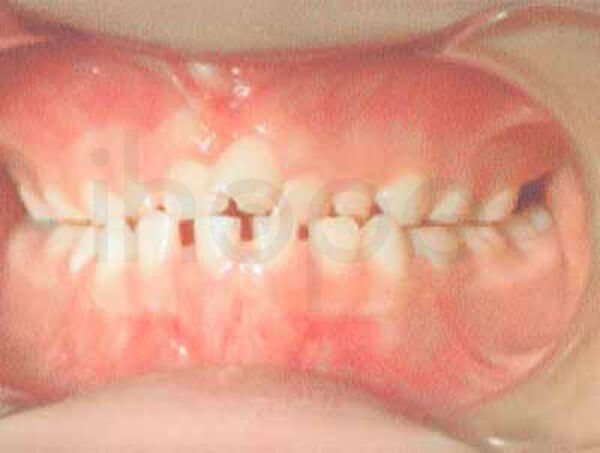
Ảnh: Thừa răng
Nguồn: National Human Genome Research Institute
Phần lớn người bệnh mắc phải chứng rụng tóc. Nam giới bị hói sau tuổi dậy thì. Da của trẻ nhão nhưng căng dần theo thời gian.
Độ phổ biến
Hội chứng Trichorhinophalangeal loại I hiếm gặp, do đó hiện nay tỷ lệ mắc bệnh vẫn chưa được thống kê cụ thể. Tại Hà Lan, khoảng 35 người mắc bệnh được báo cáo.
Nguyên nhân
Đột biến gen TRPS1 gây ra hội chứng Trichorhinophalangeal loại I.
Gen TRPS1 cung cấp hướng dẫn tạo ra một loại protein trong nhân tế bào, nó tương tác với một số vùng ADN nhằm ức chế hoạt động của các gen tương ứng. Nhiều nghiên cứu cho thấy protein TRPS1 điều chỉnh các gen kiểm soát quá trình phát triển của mô xương và xương.
Đột biến gen TRPS1 dẫn đến phiên bản protein TRPS1 giảm khả năng kiểm soát hoạt động của các gen liên quan. Do đó, xương ngón tay, ngón chân và khớp bất thường, các đặc điểm khuôn mặt khác biệt.
Trường hợp mất gen TRPS1 và các gen lân cận gây ra hội chứng Trichohinophalangeal loại II, người bệnh biểu hiện nhiều dấu hiệu và triệu chứng tương tự hội chứng Trichorhinophalangeal loại I. Ngoài ra, người bệnh còn có nhiều khối u xương sụn (không phải ung thư) và thiểu năng trí tuệ do mất một số gen gần TRPS1.
Chẩn đoán
Chẩn đoán hội chứng Trichorhinophalangeal loại I cần được đánh giá kỹ lưỡng các biểu hiện lâm sàng và tiền sử bệnh cá nhân, gia đình. Bác sĩ có thể chỉ định một số xét nghiệm hình ảnh (X-quang, MRI) nhằm đánh giá những bất thường về xương hoặc xét nghiệm di truyền tìm đột biến gen TRPS1.
Điều trị
Hiện nay chưa có phương pháp điều trị hội chứng Trichorhinophalangeal loại I. Các liệu pháp hướng đến triệu chứng cụ thể của mỗi người bệnh.
Dạng di truyền
Bệnh di truyền theo kiểu trội nhiễm sắc thể thường. Một bản sao của gen đột biến trong mỗi tế bào đủ để gây ra bệnh. Phần lớn người bệnh thừa hưởng đột biến từ cha hoặc mẹ bị bệnh.

Nguồn: U.S. National Library of Medicine
Một số trường hợp do đột biến gen mới và xảy ra ở gia đình không có tiền sử bệnh.
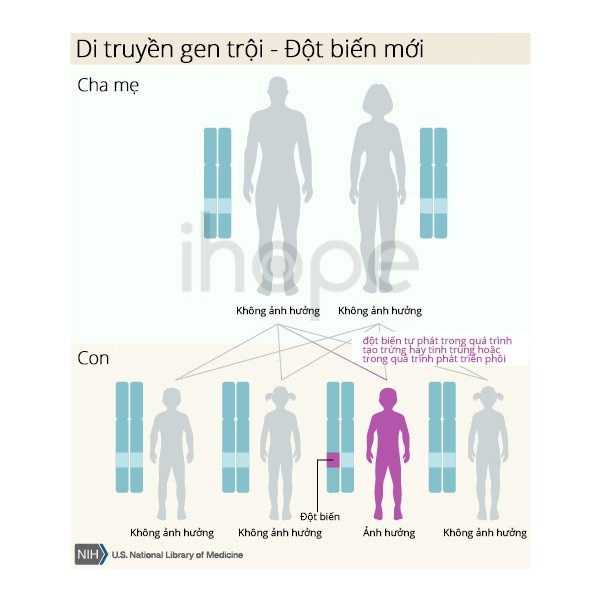
Nguồn: U.S. National Library of Medicine
Phòng ngừa
Bệnh di truyền trội trên nhiễm sắc thể thường, nếu cha hoặc mẹ mắc bệnh, con sinh ra sẽ có 50% khả năng di truyền bệnh. Do đó, để bảo đảm 100% khả năng con không bị bệnh, cha mẹ có thể chọn phương pháp thụ tinh nhân tạo IVF và sàng lọc phôi PGS/PGD. Các thành viên trong gia đình nên khám sức khỏe và tầm soát bệnh định kỳ nếu có thành viên mắc bệnh. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh.
Các tên gọi khác
- Trichorhinophalangeal dysplasia type I
- TRP syndrome
- TRPS I
- TRPS1
References
- Genetic Testing Information. Trichorhinophalangeal dysplasia type I. Retrieved January 27, 2023 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0432233/
- Genetic and Rare Diseases Information Center. Trichorhinophalangeal dysplasia type 1. Retrieved January 27, 2023 from https://rarediseases.info.nih.gov/diseases/7800/trichorhinophalangeal-syndrome-type-1
- Catalog of Genes and Diseases from OMIM. TRICHORHINOPHALANGEAL SYNDROME, TYPE I. Retrieved January 27, 2023 from https://omim.org/entry/190350
- U.S National Library of Medicine. Trichorhinophalangeal dysplasia type I. Retrieved January 27, 2023 from https://medlineplus.gov/genetics/condition/trichorhinophalangeal-syndrome-type-i/
- National Organization for Rare Disorders. Trichorhinophalangeal dysplasia type I. Retrieved January 27, 2023 from https://rarediseases.org/rare-diseases/trichorhinophalangeal-syndrome-type-i/
- Orphanet. Trichorhinophalangeal syndrome type 1 and 3. Retrieved January 27, 2023 from https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=77258
- Candamourty R, Venkatachalam S, Karthikeyan B, Babu MR. Trichorhinophalangeal syndrome type 1: A case report with literature review. J Nat Sci Biol Med. 2012 Jul;3(2):209-11. doi: 10.4103/0976-9668.101936
- Dias C, Isidoro L, Santos M, Santos H, Marques JS. Trichorhinophalangeal Syndrome Type I: A Patient with Two Novel and Different Mutations in the TRPS1 Gene. Case Rep Genet. 2013;2013:748057. doi: 10.1155/2013/748057
- Maas S, Shaw A, Bikker H, Hennekam RCM. Trichorhinophalangeal Syndrome. 2017 Apr 20. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from http://www.ncbi.nlm.nih.gov/books/NBK425926/
- Maas SM, Shaw AC, Bikker H, Ludecke HJ, van der Tuin K, Badura-Stronka M, Belligni E, Biamino E, Bonati MT, Carvalho DR, Cobben J, de Man SA, Den Hollander NS, Di Donato N, Garavelli L, Gronborg S, Herkert JC, Hoogeboom AJ, Jamsheer A, Latos-Bielenska A, Maat-Kievit A, Magnani C, Marcelis C, Mathijssen IB, Nielsen M, Otten E, Ousager LB, Pilch J, Plomp A, Poke G, Poluha A, Posmyk R, Rieubland C, Silengo M, Simon M, Steichen E, Stumpel C, Szakszon K, Polonkai E, van den Ende J, van der Steen A, van Essen T, van Haeringen A, van Hagen JM, Verheij JB, Mannens MM, Hennekam RC. Phenotype and genotype in 103 patients with tricho-rhino-phalangeal syndrome. Eur J Med Genet. 2015 May;58(5):279-92. doi: 10.1016/j.ejmg.2015.03.002.





