
Hội chứng Andersen-Tawil (Andersen-Tawil syndrome) là bệnh di truyền gây ra các cơn yếu cơ (liệt theo chu kỳ), thay đổi nhịp tim (loạn nhịp) và nhiều bất thường về phát triển. Liệt chu kỳ thường khởi phát sớm trong giai đoạn trẻ nhỏ, chúng có thể kéo dài từ vài giờ đến vài ngày và phần lớn không xác định được nguyên nhân. Triệu chứng có thể xảy ra sau khi bệnh nhân hoạt động thể lực hoặc sau những khoảng thời gian nghỉ ngơi dài. Sau những cơn tái phát, sức mạnh cơ bắp trở lại trạng thái bình thường. Một vài trường hợp có thể bị yếu cơ nhẹ vĩnh viễn.
Biểu hiện lâm sàng
Người mắc hội chứng Andersen–Tawil thường gặp phải rối loạn nhịp thất và hội chứng QT dài. Hội chứng QT dài khiến cơ tim mất nhiều thời gian hơn để khôi phục lại năng lượng giữa các chu kỳ đập. Những bất thường này dẫn đến đánh trống ngực và ngất xỉu, nghiêm trọng có thể gây tử vong.
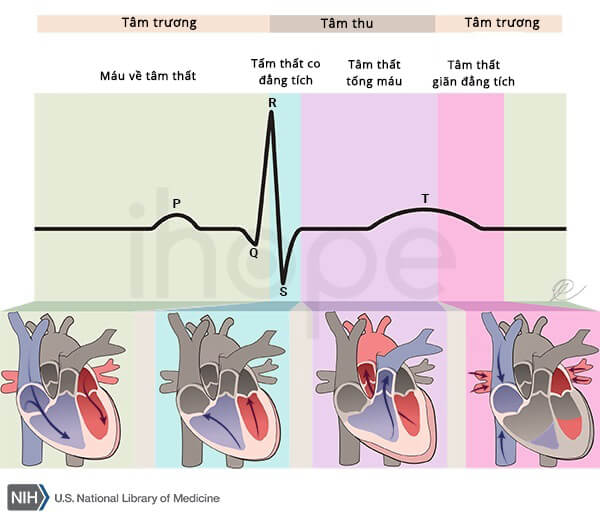
Nguồn: U.S National Library of Medicine
Ngoài ra, bệnh nhân cũng thường phát triển những dị tật về hình thái cơ thể. Những khuyết tật phổ biến bao gồm hàm nhỏ, răng mọc lệch, vị trí tai thấp, khoảng cách mắt cách, tật dính khớp ngón chân và khớp ngón tay cong lệch. Một số trường hợp mắc chứng vẹo cột sống hoặc tầm vóc thấp bé. Mức độ nghiêm trọng của những biểu hiện này khác nhau giữa mỗi bệnh nhân, ngay cả giữa các thành viên trong gia đình. Khoảng 60% số người mắc bệnh có cả ba đặc điểm chính của bệnh gồm liệt chu kì, rối loạn nhịp tim và các bất thường về thể chất.
Độ phổ biến
Hội chứng Andersen-Tawil là bệnh di truyền di truyền hiếm gặp. Hiện nay, người ta ước tính tỉ lệ mắc bệnh khoảng 1/1.000.000 người trên toàn thế giới. Hơn 200 trường hợp mắc bệnh đã được ghi nhận. Người ta cho rằng hội chứng Andersen-Tawil là nguyên nhân của 10% tổng số ca liệt chu kì.
Nguyên nhân
Đột biến gen KCNJ2 gây ra hơn 60% số trường hợp mắc hội chứng Andersen-Tawil. Bệnh nhân mang đột biến này được phân vào loại 1 (ATS1).
Gen KCNJ2 cung cấp hướng dẫn tạo ra các kênh vận chuyển ion kali mang điện tích dương qua màng tế bào cơ. Quá trình di chuyển của ion kali qua những kênh này rất quan trọng để duy trì chức năng bình thường của cơ vân và cơ tim.
Đột biến gen KCNJ2 làm thay đổi cấu trúc và chức năng của kênh kali nên dòng chảy ion kali trong cơ xương và cơ tim trở nên bất thường rồi dẫn đến biểu hiện liệt chu kỳ và rối loạn nhịp tim đặc trưng của hội chứng Andersen-Tawil. Người ta chưa xác định được vai trò của gen KCNJ2 trong quá trình phát triển của xương và cũng chưa xác định được cơ chế gây ra những thay đổi về xương và các dị tật thể chất khác của đột biến này.
Trong 40% số trường hợp không do đột biến gen KCNJ2, nguyên nhân dẫn đến hội chứng Andersen-Tawil vẫn chưa được xác định. Những trường hợp này được xếp vào loại 2 (ATS2). Các nghiên cứu cho thấy đột biến trên ít nhất một gen khác liên quan đến kênh kali có thể là nguyên nhân gây ra bệnh.
Chẩn đoán
Bác sĩ chẩn đoán hội chứng Andersen-Tawil dựa trên các biểu hiện lâm sàng (liệt chu kì, rối loạn nhịp tim có triệu chứng và các đặc điểm dị biệt trên khuôn mặt), tiền sử bệnh trong gia đình, khám sức khỏe toàn diện và một loạt xét nghiệm chuyên biệt.
Do nồng độ kali trong máu có thể giảm trong những đợt liệt chu kì, xét nghiệm máu nhằm xác định nồng độ kali trong huyết thanh có thể cung cấp thêm thông tin hỗ trợ chẩn đoán.
Xét nghiệm dẫn truyền thần kinh vận động kéo dài (long exercise nerve conduction studies) cũng có thể được sử dụng. Trong xét nghiệm này, bệnh nhân sẽ thực hiện co một cơ nhỏ phía ngoài lòng bàn tay trong khoảng 2–5 phút. Thủ thuật này cho phép bác sĩ đánh giá chức năng cơ và ghi nhận dấu hiệu của liệt chu kì.
Điện tâm đồ ghi lại dấu hiệu điện sinh lý của tim và phát hiện các mẫu hoạt động điện thường gặp trong hội chứng Andersen-Tawil. Một số bệnh nhân có thể được chẩn đoán bằng cách đo Holter 24 giờ. Thông qua các điện cực dán trên ngực, thiết bị đo liên tục ghi nhận nhịp tim nhằm phát hiện sự hiện diện, tần suất và thời lượng của các đợt loạn nhịp thất và các triệu chứng khác. Xét nghiệm di truyền phân tử có thể được sử dụng nhằm xác nhận kết quả chẩn đoán bệnh trong một số trường hợp.
Điều trị
Hiện nay, chưa có bất kỳ phác đồ điều trị đặc hiệu cho những người mắc hội chứng Andersen-Tawil. Do bệnh hiếm gặp, chưa có thử nghiệm điều trị nào được thực hiện trên một nhóm bệnh nhân lớn. Các phương pháp điều trị khác nhau được áp dụng cho các trường hợp đơn lẻ hoặc chuỗi ca bệnh nhỏ.
Những người mắc bệnh được khuyến cáo nên tránh các yếu tố gây khởi phát liệt chu kỳ như nghỉ ngơi ngay sau khi hoạt động thể chất hoặc tập thể dục quá lâu. Bên cạnh đó, người bệnh cũng nên tránh sử dụng các loại thuốc có thể kéo dài khoảng QT.
Khi liệt chu kỳ liên quan đến nồng độ kali thấp, bổ sung kali qua đường uống có thể mang lại hiệu quả. Đối với những người có nguy cơ hạ kali, bổ sung kali hàng ngày cũng nên được cân nhắc.
Một cơn liệt chu kỳ xảy ra khi nồng độ kali cao thường tự khỏi trong vòng 60 phút. Người bệnh nên ăn các loại thực phẩm giàu carbohydrate hoặc tiếp tục tập luyện nhẹ có thể rút ngắn thời gian liệt chu kì.
Dạng di truyền
Hội chứng Andersen-Tawil di truyền theo kiểu trội trên nhiễm sắc thể thường. Do đó, một bản sao của gen trong mỗi tế bào đủ để gây ra bệnh. Người bệnh thường được di truyền từ cha hoặc mẹ mắc bệnh. Một số ít trường hợp là do đột biến mới (denovo) xuất hiện trong quá trình hình thành tế bào sinh sản (trứng, tinh trùng) hoặc quá trình phát triển phôi sớm.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Hội chứng Andersen-Tawil di truyền trội trên nhiễm sắc thể thường, nếu cha hoặc mẹ mắc bệnh, con sinh ra sẽ có 50% khả năng di truyền bệnh. Do đó, để bảo đảm 100% khả năng con không bị bệnh, cha mẹ có thể chọn phương pháp thụ tinh nhân tạo IVF và sàng lọc phôi PGS/PGD. Các thành viên trong gia đình nên khám sức khỏe và tầm soát bệnh định kỳ nếu có thành viên mắc bệnh. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh.
Các tên gọi khác
- Andersen syndrome
- ATS
- Long QT syndrome 7
- LQT
References
- Genetic Testing Information. Andersen Tawil syndrome. Retrieved May 31, 2024 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C1563715/?_ga=2.221884048.980275174.1717136825-1635318420.1684225451
- Genetic and Rare Diseases Information Center. Andersen-Tawil syndrome. Retrieved May 31, 2024 from https://rarediseases.info.nih.gov/diseases/9453/index
- Catalog of Genes and Diseases from OMIM. ANDERSEN CARDIODYSRHYTHMIC PERIODIC PARALYSIS. Retrieved May 31, 2024 from https://omim.org/entry/170390
- U.S National Library of Medicine. Andersen-Tawil syndrome. Retrieved May 31, 2024 from https://medlineplus.gov/genetics/condition/andersen-tawil-syndrome/
- Frontiers. Andersen–Tawil Syndrome With Novel Mutation in KCNJ2: Case Report. Retrieved May 31, 2024 from https://www.frontiersin.org/articles/10.3389/fped.2021.790075/full
- National Organization for Rare Disorders. Andersen-Tawil Syndrome. Retrieved May 31, 2024 from https://omim.org/entry/170390
- Orphanet. Andersen-Tawil syndrome. Retrieved May 31, 2024 from https://www.orpha.net/en/disease/detail/37553
- National Library of Medicine. Andersen Syndrome: The Newest Variant of the Hereditary‐Familial Long QT Syndrome. Retrieved May 31, 2024 from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6932481/
- National Library of Medicine. Andersen-Tawil Syndrome: A Comprehensive Review. Retrieved May 31, 2024 from https://pubmed.ncbi.nlm.nih.gov/32947483/





