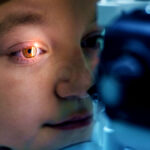
Không mống mắt (aniridia) là bệnh di truyền ảnh hưởng đến mắt. Người bệnh thường thiếu hoàn toàn hoặc một phần mống mắt (phần có màu của mắt). Những bất thường tại mống mắt có thể khiến đồng tử biến dạng hoặc không bình thường.
Biểu hiện lâm sàng
Người bệnh thường bị giảm thị lực và tăng độ nhạy cảm với ánh sáng. Ngoài ra, áp lực trong nhãn cầu tăng cao (bệnh tăng nhãn áp) thường xuất hiện vào cuối giai đoạn trẻ nhỏ hoặc đầu tuổi dậy thì. Khoảng 50–85% người bệnh bị đục thủy tinh thể. Khoảng 10% người bệnh có dây thần kinh thị giác (cấu trúc truyền thông tin từ mắt đến não) phát triển không đầy đủ. Họ có thể bị rung giật nhãn cầu không kiểm soát hoặc vùng võng mạc trung tâm phụ trách thị lực chính xác phát triển kém. Những vấn đề này góp phần làm thị lực ngày càng suy giảm. Mức độ nghiêm trọng của các triệu chứng thường giống nhau trên cả hai mắt.
Trong một số trường hợp hiếm gặp, người bệnh có thể gặp rối loạn hành vi, chậm phát triển và khó nhận biết mùi.
Độ phổ biến
Tỉ lệ mắc không mống mắt khoảng 1/50.000–100.000 trẻ sơ sinh trên toàn thế giới.
Nguyên nhân
Đột biến gen PAX6 là nguyên nhân gây nên bệnh không mống mắt. Gen PAX6 cung cấp hướng dẫn tạo ra protein tham gia vào giai đoạn phát triển ban đầu của mắt, não và tủy sống (hệ thần kinh trung ương) cùng với tuyến tụy. Trong não, protein PAX6 góp phần phát triển nhóm tế bào não chuyên biệt xử lí khứu giác (hành khứu). Protein PAX6 gắn kết với một số vùng ADN nhằm điều hòa hoạt động của các gen tương ứng, do đó nó được gọi protein điều hòa phiên mã. Sau khi sinh, protein PAX6 điều hòa nhiều gen góp phần duy trì các cấu trúc khác nhau của mắt.
Đột biến gen PAX6 tạo ra protein không hoạt động, do đó protein không thể gắn kết với ADN để điều hòa hoạt động của các gen khác. Do đó, quá trình hình thành mắt bị gián đoạn trong thời kì phát triển phôi thai.
Chẩn đoán
Nhằm chẩn đoán bệnh không mống mắt, bác sĩ thường thực hiện nhiều xét nghiệm khác nhau. Đầu tiên, chụp mạch huỳnh quang đáy mắt được thực hiện nhằm quan sát vùng trung tâm võng mạc. Tiếp theo, chụp OCT để đánh giá cấu trúc võng mạc, chủ yếu là hố trung tâm. Đối với trẻ mắc không mống mắt bẩm sinh đơn lẻ, trẻ cần định kì siêu âm bụng nhằm phát hiện sớm khối u Wilms—một biến chứng nguy hiểm có thể xảy ra. Bác sĩ cũng có thể chỉ định xét nghiệm gen PAX6 nhằm xác định nguyên nhân di truyền. Ngoài ra, các thành viên trong gia đình người bệnh nên khám mắt nhằm tìm kiếm các trường hợp mắc bệnh chưa được phát hiện.
Điều trị
Quá trình điều trị bệnh không mống mắt cần được thực hiện sớm và toàn diện. Trẻ nhỏ mắc bệnh cần được đeo kính nhằm cải thiện thị lực, kết hợp điều trị nhược thị và chỉnh lác nếu có. Để giảm nhạy cảm với ánh sáng và cải thiện thẩm mĩ, bệnh nhân có thể đeo kính áp tròng có màu hoặc kính mắt đổi màu. Trong những trường hợp nặng, bệnh nhân có thể cân nhắc cấy mống mắt nhân tạo.
Các biến chứng của không mống mắt cũng cần được điều trị tích cực. Với bệnh tăng nhãn áp, người bệnh cần can thiệp phẫu thuật vì thuốc ít hiệu quả. Các phương pháp phẫu thuật bao gồm mổ góc tiền phòng, cắt bè hoặc đặt van dẫn lưu. Đục thủy tinh thể có thể cần phẫu thuật thay thế, tuy nhiên đây là ca phẫu thuật phức tạp do cấu trúc mắt yếu. Với tổn thương giác mạc, tùy mức độ mà có thể điều trị bằng thuốc nhỏ mắt, ghép màng ối hoặc ghép giác mạc. Ngoài ra, các vấn đề như lác và sụp mi cũng cần được điều trị kĩ lưỡng.
Dạng di truyền
Bệnh không mống mắt di truyền trội trên nhiễm sắc thể thường, nghĩa là một bản sao của gen đột biến trong mỗi tế bào là đủ để gây ra bệnh. Phần lớn người bệnh đều có cha mẹ và các thành viên khác trong gia đình mắc bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Bệnh không mống mắt di truyền trội trên nhiễm sắc thể thường, nếu cha hoặc mẹ mắc bệnh, con sinh ra sẽ có 50% khả năng di truyền bệnh. Do đó, để bảo đảm 100% khả năng con không bị bệnh, cha mẹ có thể chọn phương pháp thụ tinh nhân tạo IVF và sàng lọc phôi PGS/PGD. Các thành viên trong gia đình nên khám sức khỏe và tầm soát bệnh định kì nếu có thành viên mắc bệnh. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh.
Các tên gọi khác
- Absent iris
- Congenital aniridia
- Irideremia
References
- National Library of Medicine. Congenital aniridia. Retrieved June 22, 2025 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0003076/
- Genetic and Rare Diseases Information Center. Isolated aniridia. Retrieved June 22, 2025 from https://rarediseases.info.nih.gov/diseases/5816/index
- OMIM. ANIRIDIA 1; AN1. Retrieved June 22, 2025 from https://omim.org/entry/106210
- MedlinePlus. Aniridia. Retrieved June 22, 2025 from https://medlineplus.gov/genetics/condition/aniridia/
- StatPearls [Internet]. Aniridia. Retrieved June 22, 2025 from https://www.ncbi.nlm.nih.gov/books/NBK538133/