
Loạn dưỡng cơ bẩm sinh Fukuyama (Fukuyama congenital muscular dystrophy) là một trong nhóm các bệnh loạn dưỡng cơ bẩm sinh gây yếu và teo cơ. Triệu chứng của bệnh thường khởi phát sớm ngay sau sinh và tiến triển chậm trong giai đoạn tiếp theo khiến trẻ mắc các khiếm khuyết về cơ, não và mắt.
Nguồn: U.S. National Library of Medicine.
Biểu hiện lâm sàng
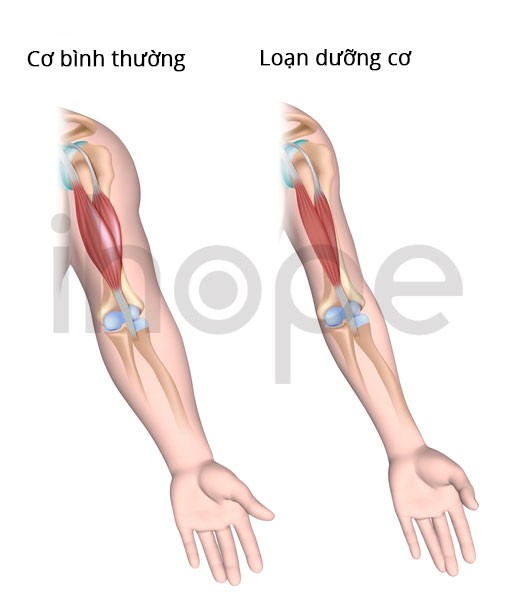
Loạn dưỡng cơ bẩm sinh Fukuyama ảnh hưởng đến các cơ tham gia vào quá trình vận động (cơ xương). Các dấu hiệu đầu tiên của bệnh xuất hiện ngay trong giai đoạn sơ sinh bao gồm khóc yếu, bú kém và lực cơ giảm. Cơ mặt yếu thường dẫn đến các đặc điểm dị biệt trên khuôn mặt bao gồm mí mắt sụp xuống (sụp mi) và miệng há hốc. Trong giai đoạn trẻ nhỏ, tình trạng yếu cơ và biến dạng khớp gây hạn chế vận động và cản trở quá trình phát triển các kỹ năng vận động như ngồi, đứng và đi lại.
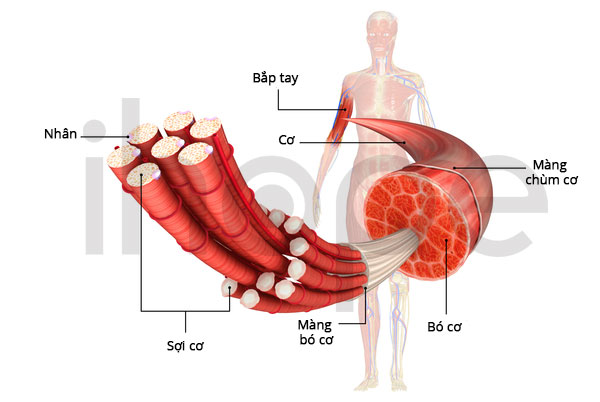
Ảnh: Giải phẫu cơ
Nguồn: Medlineplus.gov
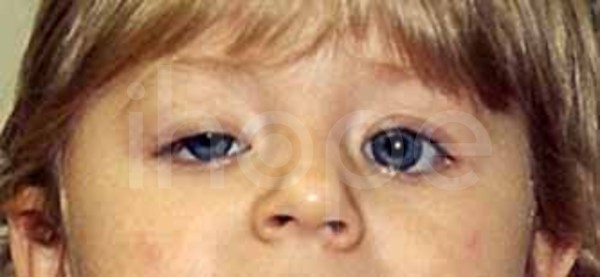
Ảnh: Mí mắt sụp xuống
Nguồn: Elements of Morphology, National Human Genome Research Institute
Loạn dưỡng cơ bẩm sinh Fukuyama cũng làm suy giảm quá trình phát triển của não, từ đó gây ra bệnh sỏi lissencephaly (cobblestone lissencephaly) với bề mặt não có hình dạng gập ghềnh và không đều (giống như đá cuội). Những thay đổi trong cấu trúc của não khiến trẻ chậm phát triển kỹ năng nói, vận động cũng như chậm phát triển trí tuệ từ mức độ trung bình đến nặng. Các kỹ năng xã hội thường ít chịu tác động hơn. Phần lớn trẻ mắc loạn dưỡng cơ bẩm sinh Fukuyama không thể tự đứng hoặc đi lại, một số trường hợp trẻ có thể ngồi mà không cần hỗ trợ. Ngoài ra, hơn một nửa số trẻ mắc kèm bệnh động kinh.
Các dấu hiệu và triệu chứng khác của loạn dưỡng cơ bẩm sinh Fukuyama bao gồm:
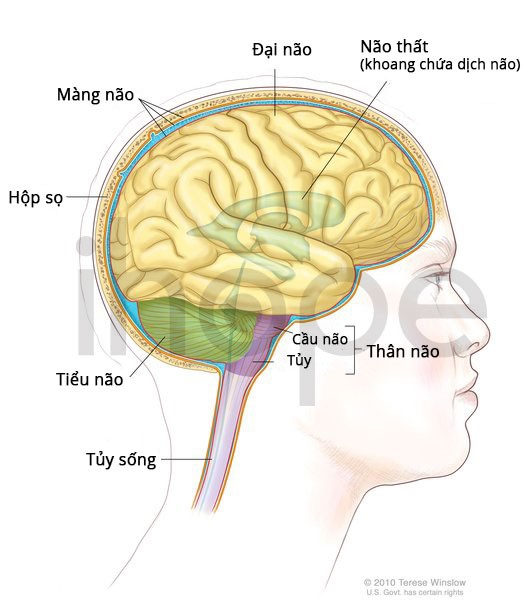
Ảnh: Cấu trúc bên trong của não
Nguồn: Terese Winslow
- Suy giảm thị lực
- Bất thường mắt
- Khó nuốt
- Nhiễm trùng
- Bệnh tim tiến triển chậm sau 10 tuổi
Do loạn dưỡng cơ bẩm sinh Fukuyama gây ra các vấn đề nghiêm trọng đối với sức khỏe, đa số những người mắc bệnh chỉ sống được đến cuối giai đoạn trẻ nhỏ hoặc thanh thiếu niên.
Độ phổ biến
Loạn dưỡng cơ bẩm sinh Fukuyama phần lớn được tìm thấy tại Nhật Bản với tỉ lệ mắc bệnh là 2-4/100.000 trẻ sơ sinh. Đây là dạng loạn dưỡng cơ phổ biến thứ hai trên trẻ (sau loạn dưỡng cơ Duchenne).
Nguyên nhân
Đột biến gen FKTN gây ra bệnh loạn dưỡng cơ bẩm sinh Fukuyama. Gen này cung cấp hướng dẫn tạo ra protein fukutin có chức năng neo các tế bào vào chất nền ngoại bào—một mạng lưới protein và phân tử bao quanh bên ngoài tế bào. Mặc dù chức năng chính xác của fukutin vẫn chưa rõ ràng, người ta dự đoán protein này có thể biến đổi alpha (α)-dystroglycan về mặt hóa học. Trong cơ xương, α-dystroglycan có chức năng ổn định và bảo vệ các sợi cơ. Trong não, protein này định hướng tế bào thần kinh di chuyển trong quá trình phát triển ban đầu.
Đột biến gen FKTN làm giảm lượng fukutin trong tế bào, từ đó gây gián đoạn quá trình biến đổi α-dystroglycan thành phân tử có chức năng, nên tế bào cơ không được bảo vệ sẽ dễ tổn thương khi chúng co giãn liên tục. Cuối cùng, sợi cơ yếu đi và chết dần theo thời gian, triệu chứng yếu cơ từ đó khởi phát.
Khiếm khuyết α-dystroglycan cũng ảnh hưởng đến hoạt động di chuyển của các tế bào thần kinh trong giai đoạn phát triển ban đầu của não. Thông thường, tế bào thần kinh sẽ dừng lại khi đến đúng vị trí mục tiêu, tuy nhiên khiếm khuyết này khiến một số tế bào di chuyển qua bề mặt não đến vùng không gian chứa đầy chất lỏng bao quanh. Người ta cho rằng quá trình di chuyển tế bào thần kinh gặp bất thường gây ra chứng sỏi lissencephaly trên trẻ mắc loạn dưỡng cơ bẩm sinh Fukuyama. Hiện nay, người ta vẫn chưa hiểu rõ cơ chế tác động của đột biến gen FNTK lên các bộ phận khác trên cơ thể.
Loạn dưỡng cơ bẩm sinh Fukuyama còn được mô tả là chứng dystroglycanopathy do chúng liên quan đến một sai hỏng của α-dystroglycan.
Chẩn đoán
Bác sĩ chẩn đoán loạn dưỡng cơ bẩm sinh Fukuyama thông qua khám sức khỏe toàn diện và tiền sử bệnh. Ngoài ra, xét nghiệm máu có thể được chỉ định nhằm phát hiện nồng độ enzyme creatine kinase cao bất thường được giải phóng từ tế bào cơ bị tổn thương. Xét nghiệm di truyền tìm đột biến gen FKTN và đo điện cơ xác định vùng cơ tổn thương cũng được ứng dụng trong chẩn đoán bệnh. Ngoài ra, bác sĩ có thể tiến hành sinh thiết cơ và xét nghiệm nhằm phân biệt loạn dưỡng cơ với các bệnh thần kinh cơ khác.
Điều trị
Hiện nay, chưa có phương pháp điều trị hoàn toàn bệnh loạn dưỡng cơ bẩm sinh Fukuyama. Các liệu pháp được sử dụng chủ yếu giảm nhẹ triệu chứng và cải thiện chất lượng đời sống bệnh nhân. Đối với những bệnh nhân có triệu chứng động kinh, các loại thuốc có thể đề xuất bao gồm phenytoin, axit valproic, phenobarbitol, clonazepam, ethusuximide, primidone, corticotropin và thuốc corticosteroid. Bệnh nhân có thể được điều trị bằng vật lý trị liệu nhằm ngăn ngừa cứng khớp. Ngoài ra, bệnh nhân và gia đình nên được tư vấn đầy đủ về di truyền.
Dạng di truyền
Loạn dưỡng cơ bẩm sinh Fukuyama di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh lặn trên nhiễm sắc thể thường sẽ có bố và mẹ mang một bản sao của gen đột biến, nhưng bố mẹ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Trường hợp Loạn dưỡng cơ bẩm sinh Fukuyama di truyền lặn do đột biến gen FKTN, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kỳ.
Các tên gọi khác
- Cerebromuscular dystrophy, Fukuyama type
- FCMD
- Fukuyama CMD
- Fukuyama muscular dystrophy
- Fukuyama syndrome
- Fukuyama type congenital muscular dystrophy
- Muscular dystrophy, congenital progressive, with mental retardation
- Muscular dystrophy, congenital, Fukuyama type
- Muscular dystrophy, congenital, with central nervous system involvement
- Polymicrogyria with muscular dystrophy
References
- Genetic Testing Information. Muscular dystrophy-dystroglycanopathy (congenital with brain and eye anomalies), type A, 4. Retrieved November 27, 2023 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C2745959/
- Genetic and Rare Diseases Information Center. Congenital muscular dystrophy, Fukuyama type. Retrieved November 27, 2023 from https://rarediseases.info.nih.gov/diseases/6475/fukuyama-type-muscular-dystrophy
- Catalog of Genes and Diseases from OMIM. MUSCULAR DYSTROPHY-DYSTROGLYCANOPATHY (CONGENITAL WITH BRAIN AND EYE ANOMALIES), TYPE A, 4; MDDGA4. Retrieved November 27, 2023 from https://omim.org/entry/253800
- MedlinePlus. Fukuyama congenital muscular dystrophy. Retrieved November 27, 2023 from https://medlineplus.gov/genetics/condition/fukuyama-congenital-muscular-dystrophy/
- National Organization for Rare Disorders. Fukuyama Type Congenital Muscular Dystrophy. Retrieved November 27, 2023 from https://rarediseases.org/rare-diseases/fukuyama-type-congenital-muscular-dystrophy/




