Thiếu pyruvate carboxylase là bệnh di truyền khiến axit lactic và các hợp chất gây độc khác tích tụ trong máu đến mức làm hỏng các cơ quan và mô của cơ thể, chủ yếu hệ thần kinh.
Nguồn: Eunice Kennedy Shriver National Institute of Child Health and Human Development
Biểu hiện lâm sàng
Bệnh khiến các tế bào máu vỡ ra rồi giải phóng bilirubin. Nồng độ bilirubin cao gây ra chứng vàng da.
Thiếu pyruvate carboxylase được chia làm 3 loại chính dựa trên mức độ nghiêm trọng và triệu chứng.
Thiếu pyruvate carboxylase loại A khởi phát các biểu hiện nghiêm trọng từ giai đoạn trẻ nhỏ, người bệnh chủ yếu có gốc Bắc Mỹ. Trẻ chậm phát triển và tích tụ axit lactic trong máu (nhiễm axit lactic). Nhiều axit lactic trong máu gây ra nhiều triệu chứng như nôn mửa, đau bụng, mệt mỏi, yếu cơ và khó thở. Trong một số trường hợp, các triệu chứng xuất hiện sau giai đoạn nhịn ăn hoặc mắc bệnh khác. Người thiếu pyruvate carboxylase loại A thường không sống sót qua giai đoạn sơ sinh hoặc trẻ nhỏ.
Thiếu pyruvate carboxylase loại B có triệu chứng nghiêm trọng đe dọa tính mạng và xuất hiện ngay sau sinh. Những trường hợp mắc bệnh chủ yếu được ghi nhận tại châu Âu, nhiều nhất tại Pháp. Trẻ sơ sinh bị nhiễm axit lactic nặng và amoniac tích tụ gây suy gan. Ngoài ra, bệnh nhân gặp vấn đề thần kinh bao gồm nhược cơ, cử động bất thường, co giật và hôn mê. Trẻ sơ sinh mắc loại B thường tử vong trong vòng 3 tháng sau sinh.
Thiếu pyruvate carboxylase loại C có biểu hiện nhẹ hơn những loại khác. Người bệnh thường có nồng độ axit lactic trong máu tăng nhẹ dẫn đến các vấn đề thần kinh.
Ngoài ra, thiếu pyruvate carboxylase có thể khiến thai nhi bị thiếu máu dẫn đến phù thai. Do đó, chất lỏng tích tụ nghiêm trong trong các mô và cơ quan. Tim thai nhi hoạt động quá sức nhằm đảm bảo cung cấp đầy đủ máu cho cơ thể. Thiếu máu có thể gây sinh non.
Độ phổ biến
Thiếu pyruvate carboxylase là bệnh hiếm gặp. Người ta ước tính tỉ lệ mắc bệnh khoảng 1/250.000 trẻ sơ sinh trên toàn thế giới. Loại A thường phổ biến trong một số bộ lạc da đỏ Algonkian tại miền đông Canada.
Nguyên nhân
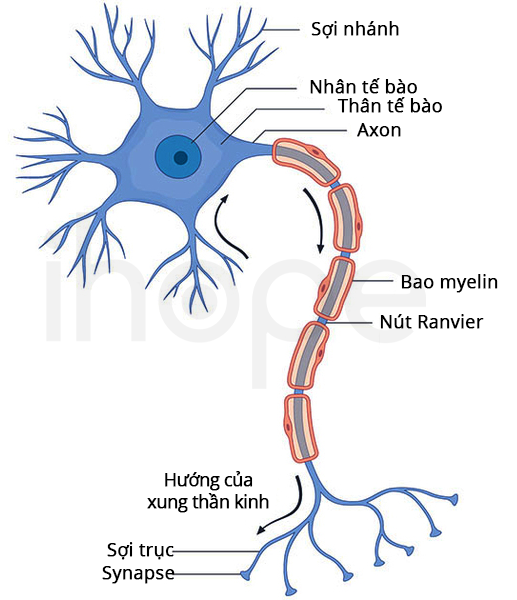
Đột biến gen PC gây ra chứng thiếu pyruvate carboxylase. Gen PC cung cấp hướng dẫn tạo ra enzyme pyruvate carboxylase. Enzyme này hiện diện chủ yếu trong ty thể—trung tâm sản xuất năng lượng của tế bào. Pyruvate carboxylase tham gia một số hoạt động quan trọng bao gồm quá trình tạo glucose. Ngoài ra, pyruvate carboxylase góp phần hình thành lớp vỏ bảo vệ quanh tế bào thần kinh (bao myelin) và chất hỗ trợ dẫn truyền thần kinh.
Đột biến gen PC khiến enzyme pyruvate carboxylase giảm số lượng hoặc mất chức năng. Do đó, quá trình tạo glucose bị gián đoạn dẫn đến ty thể suy giảm khả năng sản xuất năng lượng. Ngoài ra, thiếu pyruvate carboxylase làm axit lactic và amoniac tích tụ đến mức gây hại các cơ quan và mô. Người ta cho rằng enzyme pyruvate carboxylase giảm hoạt động trong quá trình hình thành bao myelin và chất hỗ trợ dẫn truyền thần kinh dẫn đến các vấn đề thần kinh liên quan.
Chẩn đoán
Các biểu hiện nghi ngờ thiếu pyruvate carboxylase bao gồm nôn mửa, đau bụng, mệt mỏi, axit lactic trong máu tăng và một số vấn đề thần kinh. Các xét nghiệm chuyên biệt có thể được chỉ định bao gồm kiểm tra nồng độ nồng độ axit amin, axit hữu cơ, glucose và amoniac trong huyết thanh. Khảo sát hoạt động enzyme pyruvate carboxylase trong nguyên bào sợi cho thấy hoạt tính enzyme này giảm. Ngoài ra, xét nghiệm di truyền nhằm phát hiện các đột biến gen liên quan giúp xác nhận các kết quả chẩn đoán.
Bệnh cần được chẩn đoán phân biệt với một số chứng thiếu hoạt tính enzyme khác bao gồm thiếu biotinidase, pyruvate dehydrogenase, giảm tổng hợp holocarboxylase, bất thường chuỗi hô hấp, chu trình axit tricarboxylic và khiếm khuyết tạo glucose.
Điều trị
Hiện nay chưa có phương pháp điều trị hoàn toàn chứng thiếu pyruvate carboxylase. Các liệu pháp nhằm giảm nhẹ các triệu chứng và cải thiện chất lượng đời sống bệnh nhân. Người mắc bệnh thường được cung cấp nguồn năng lượng thay thế và điều chỉnh nhiễm toan chuyển hóa cấp tính.
Dạng di truyền
Thiếu pyruvate carboxylase di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh lặn trên nhiễm sắc thể thường sẽ có bố và mẹ mang một bản sao của gen đột biến, nhưng bố mẹ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Bệnh di truyền lặn do đột biến gen PC, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kỳ.
Các tên gọi khác
- Ataxia with lactic acidosis, type II
- Leigh necrotizing encephalopathy due to pyruvate carboxylase deficiency
- Leigh syndrome due to pyruvate carboxylase deficiency
- PC deficiency
- Pyruvate carboxylase deficiency disease
- Type II ataxia with lactic acidosis
References
- Genetic Testing Information. Pyruvate carboxylase deficiency. Retrieved July 18, 2023 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0034341/
- Genetic and Rare Diseases Information Center. Pyruvate carboxylase deficiency. Retrieved July 18, 2023 from https://rarediseases.info.nih.gov/diseases/7512/pyruvate-carboxylase-deficiency
- Catalog of Genes and Diseases from OMIM. PYRUVATE CARBOXYLASE DEFICIENCY. Retrieved July 18, 2023 from https://omim.org/entry/266150
- U.S National Library of Medicine. Pyruvate carboxylase deficiency. Retrieved July 18, 2023 from https://medlineplus.gov/genetics/condition/pyruvate-carboxylase-deficiency/
- Frontiers. PC Splice-Site Variant c.1825+5G>A Caused Intron Retention in a Patient With Pyruvate Carboxylase Deficiency: A Case Report. Retrieved July 18, 2023 from https://www.frontiersin.org/articles/10.3389/fped.2022.825515/full
- MalaCards. Pyruvate Carboxylase Deficiency (PC DEFICIENCY). Retrieved July 18, 2023 from https://www.malacards.org/card/pyruvate_carboxylase_deficiency
- MSD Manuals. Pyruvate Metabolism Disorders. Retrieved July 18, 2023 from https://www.msdmanuals.com/professional/pediatrics/inherited-disorders-of-metabolism/pyruvate-metabolism-disorders
- National Organization for Rare Disorders. Pyruvate Carboxylase Deficiency. Retrieved July 18, 2023 from https://rarediseases.org/rare-diseases/pyruvate-carboxylase-deficiency/
- Orphanet. Pyruvate carboxylase deficiency. Retrieved July 18, 2023 from https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=3008