Thiếu máu nguyên hồng cầu liên-kết-X là bệnh di truyền hiếm gặp. Người bệnh bị suy giảm quá trình phát triển tế bào hồng cầu khỏe mạnh – tế bào vận chuyển oxy trong cơ thể. Do đó, tế bào hồng cầu nhỏ hơn bình thường và giảm sắc tố vì thiếu hemoglobin. Ngoài ra, sắt tích tụ bất thường xung quanh các tế bào hồng cầu tạo thành nguyên hồng cầu sắt vòng (ring sideroblast). Bệnh xảy ra phổ biến ở thanh thiếu niên.
Nguồn: Designua/Shutterstock.com
Biểu hiện lâm sàng
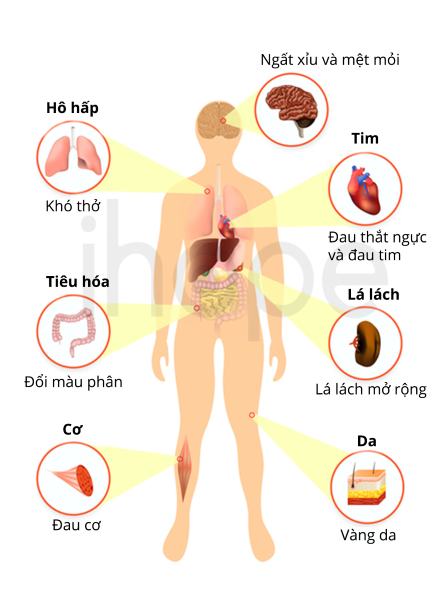
Người bệnh có các triệu chứng đặc trưng của bệnh thiếu máu bao gồm mệt mỏi, chóng mặt, tim đập nhanh, da màu đồng hoặc hơi nâu, tiểu đường mất kiểm soát, gan và lá lách to. Theo thời gian, sắt tích tụ nhiều dẫn đến các biến chứng nghiêm trọng như bệnh tim và xơ gan. Trẻ sơ sinh và trẻ nhỏ mắc bệnh có thể bị đe dọa tính mạng do quá tải sắt.
Nguồn: U.S National Library of Medicine
Độ phổ biến
Thiếu máu nguyên hồng cầu liên-kết-X hiếm gặp, do đó hiện nay tỷ lệ mắc bệnh vẫn chưa được thống kê cụ thể.
Nguyên nhân
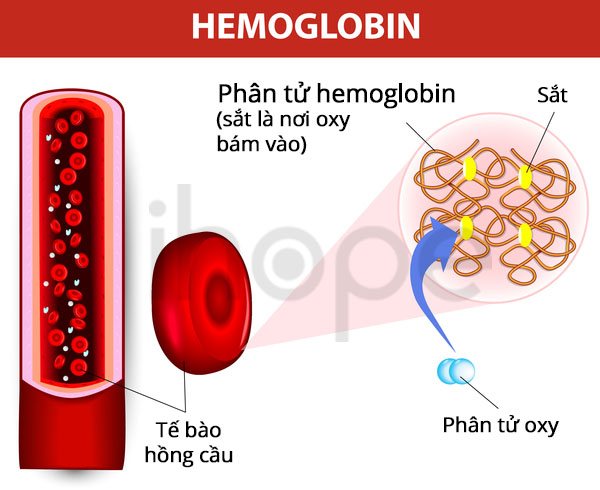
Đột biến gen ALAS2 là nguyên nhân chính gây ra bệnh thiếu máu nguyên hồng cầu liên-kết-X. Gen ALAS2 cung cấp hướng dẫn tạo ra enzyme erythroid ALA-synthase. Enzyme này nắm vai trò quan trọng trong quá trình sản xuất heme - thành phần của protein hemoglobin trong tủy xương. Đột biến gen ALAS2 làm suy giảm hoạt động của erythroid ALA-synthase, từ đó gây gián đoạn quá trình sản xuất heme và ngăn cản nguyên bào hồng cầu tạo hemoglobin. Phần lớn sắt được vận chuyển vào nguyên bào hồng cầu bằng cách gắn vào heme, do đó heme suy giảm dẫn đến tích tụ sắt dư thừa trong tế bào. Cơ thể bù đắp lượng hemoglobin bị thiếu bằng cách hấp thụ nhiều sắt hơn từ thực phẩm nên sắt dư thừa tích tụ đến mức gây hại cho các cơ quan. Nồng độ hemoglobin thấp và tích tụ sắt dẫn đến các triệu chứng đặc trưng của bệnh thiếu máu nguyên bào hồng cầu liên-kết-X.
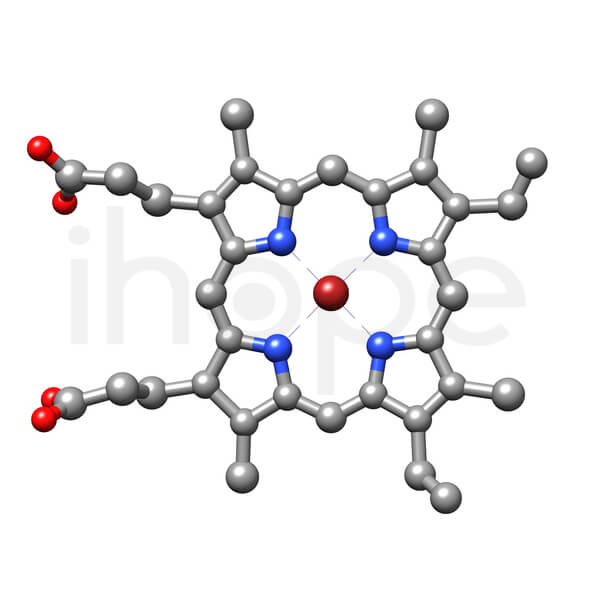
Heme là một phân tử nhỏ, phẳng với một ion sắt (màu đỏ sẫm) tại trung tâm. Nó là thành phần thiết yếu của hemoglobin, loại protein trong máu mang oxy đi khắp cơ thể.
Nguồn: Rachel Kramer Green, RCSB Protein Data Bank
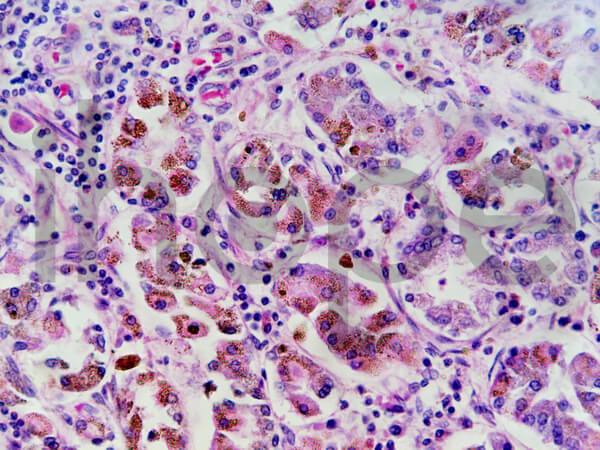
Ảnh: Sắt tích tụ trong tế bào dạ dày
Nguồn: Jubal Harshaw/Shutterstock.com
Một số người bệnh mang cả đột biến gen HFE và ALAS2 dẫn đến thiếu máu nguyên bào hồng cầu liên-kết-X dạng nghiêm trọng. Bệnh gây thừa sắt quá mức, riêng đột biến gen HFE đã làm tăng hấp thụ sắt từ thực phẩm gây ra hội chứng quá tải sắt.
Chẩn đoán
Người bệnh được khám sức khỏe, thăm hỏi tiền sử bệnh và kiểm tra các dấu hiệu lâm sàng nhằm loại trừ các bệnh lý thiếu máu có dấu hiệu tương tự. Bác sĩ có thể chỉ định một số xét nghiệm chẩn đoán bổ sung bao gồm:
- Công thức máu toàn phần (CBC) nhằm đánh giá kích thước và hình dạng của tế bào máu, số lượng tế bào hồng cầu trưởng thành và chưa trưởng thành
- Xét nghiệm máu ngoại vi
- Kiểm tra hàm lượng sắt
- Chọc hút tủy xương
- Xét nghiệm di truyền tìm đột biến gen gây bệnh

Nguồn: National Cancer Institute
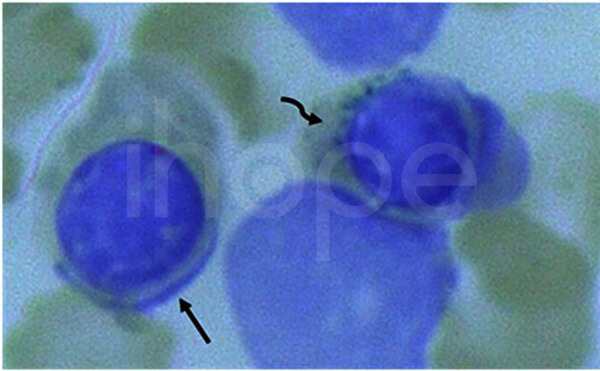
Chọc hút tủy xương và tiến hành nhuộm xanh Prussian cho thấy tiền nhân hồng cầu bình thường (mũi tên thẳng) và nguyên bào hình nhẫn chứa nhiều hạt sắt xung quanh nhân (mũi tên cong).
Nguồn: U.S National Library of Medicine
Điều trị
Hiện nay chưa có phương pháp điều trị hoàn toàn chứng thiếu máu nguyên hồng cầu liên-kết-X. Một số liệu pháp giúp giảm nhẹ triệu chứng của bệnh như bổ sung vitamin B6. Trường hợp thiếu máu nặng, người bệnh cần được truyền máu nhằm cung cấp đủ hemoglobin cho cơ thể. Sắt dư thừa có thể được loại bỏ bằng máy. Trẻ em có thể dùng thuốc thải sắt qua đường uống và đường tiêm.
Dạng di truyền
Bệnh di truyền theo kiểu lặn liên kết với nhiễm sắc thể giới tính X. Nam giới chỉ có một nhiễm sắc thể X, do đó một bản sao của gen ALAS2 đột biến trong mỗi tế bào đủ gây thiếu máu và các triệu chứng nghiêm trọng khác của bệnh. Người cha bị bệnh không truyền tính trạng này cho con trai.
Nữ giới có hai nhiễm sắc thể X, nếu trong mỗi tế bào của người phụ nữ xuất hiện một gen ALAS2 đột biến, họ là người mang gen và có thể di truyền cho thế hệ sau. Hầu hết họ không biểu hiện bất kỳ triệu chứng nào, tuy nhiên xét nghiệm máu cho thấy có các tế bào máu đỏ nhạt và nhỏ bất thường.
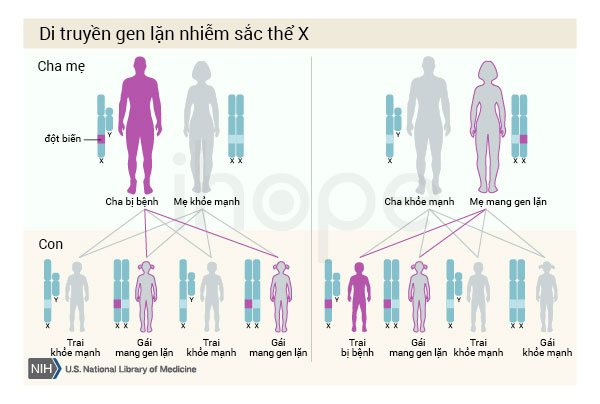
Nguồn: U.S. National Library of Medicine
Phòng ngừa
Bệnh có cơ chế di truyền liên kết X phức tạp nên khó phát hiện ở những người phụ nữ mang gen bệnh cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Do đó, các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kỳ.
Các tên gọi khác
- Anemia, hereditary sideroblastic
- Anemia, sex-linked hypochromic sideroblastic
- ANH1
- Congenital sideroblastic anaemia
- Erythroid 5-aminolevulinate synthase deficiency
- Hereditary iron-loading anemia
- X chromosome-linked sideroblastic anemia
- X-linked pyridoxine-responsive sideroblastic anemia
- XLSA
References
- Genetic Testing Information. X-linked sideroblastic anemia 1. Retrieved October 04, 2022 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C4551511/
- Genetic and Rare Diseases Information Center. Sideroblastic anemia. Retrieved October 04, 2022 from https://rarediseases.info.nih.gov/diseases/667/sideroblastic-anemia
- Genetic and Rare Diseases Information Center. X-linked sideroblastic anemia. Retrieved October 04, 2022 from https://rarediseases.info.nih.gov/diseases/9456/x-linked-sideroblastic-anemia
- Catalog of Genes and Diseases from OMIM. ANEMIA, SIDEROBLASTIC, 1. Retrieved October 04, 2022 from https://omim.org/entry/300751
- U.S National Library of Medicine. X-linked sideroblastic anemia. Retrieved October 04, 2022 from https://medlineplus.gov/genetics/condition/x-linked-sideroblastic-anemia/
- National Organization for Rare Disorders. X-linked sideroblastic anemia. Retrieved October 04, 2022 from https://rarediseases.org/gard-rare-disease/x-linked-sideroblastic-anemia/
- Cleveland Clinic. Sideroblastic Anemia. Retrieved October 04, 2022 from https://my.clevelandclinic.org/health/diseases/22932-sideroblastic-anemia
- Boston Children's Hospital. Congenital Sideroblastic Anemia. Retrieved October 04, 2022 from https://www.childrenshospital.org/conditions/congenital-sideroblastic-anemia
- Orphanet. X-linked sideroblastic anemia. Retrieved October 04, 2022 from https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=75563
- Shiqiu Xiong, Yang Jia, Shijun Li, Peng Huang, Jie Xiong, Dingan Mao, Qingnan He and Liqun Liu. The First Case Report of X-Linked Sideroblastic Anemia With Ataxia of Chinese Origin and Literature Review. Published July 20, 2021 https://doi.org/10.3389/fped.2021.692459