
Bệnh dự trữ glycogen loại VII (glycogen storage disease type VII) khiến cơ thể mất khả năng phân hủy glycogen trong tế bào cơ. Điều này cản trở các tế bào cơ thực hiện chức năng bình thường của chúng.
Biểu hiện lâm sàng
Bệnh dự trữ glycogen loại VII được phân thành bốn dạng dựa trên độ tuổi khởi phát và dấu hiệu của bệnh.
Dạng cơ bản
Bệnh dự trữ glycogen loại VII dạng cơ bản phổ biến nhất. Nó thường xuất hiện trong giai đoạn trẻ nhỏ.
Bệnh nhân xuất hiện các biểu hiện phổ biến như:
- Đau cơ và chuột rút sau mỗi lần vận động nhẹ
- Buồn nôn và nôn sau khi vận động mạnh
Trong quá trình vận động, mô cơ phân hủy bất thường giải phóng protein myoglobin. Bình thường, thận chuyển hóa protein myoglobin rồi bài tiết chúng qua nước tiểu. Tuy nhiên, thận của người bệnh không thể bài tiết mà tích trữ lượng lớn myoglobin đến mức dẫn đến tổn thương và suy thận.
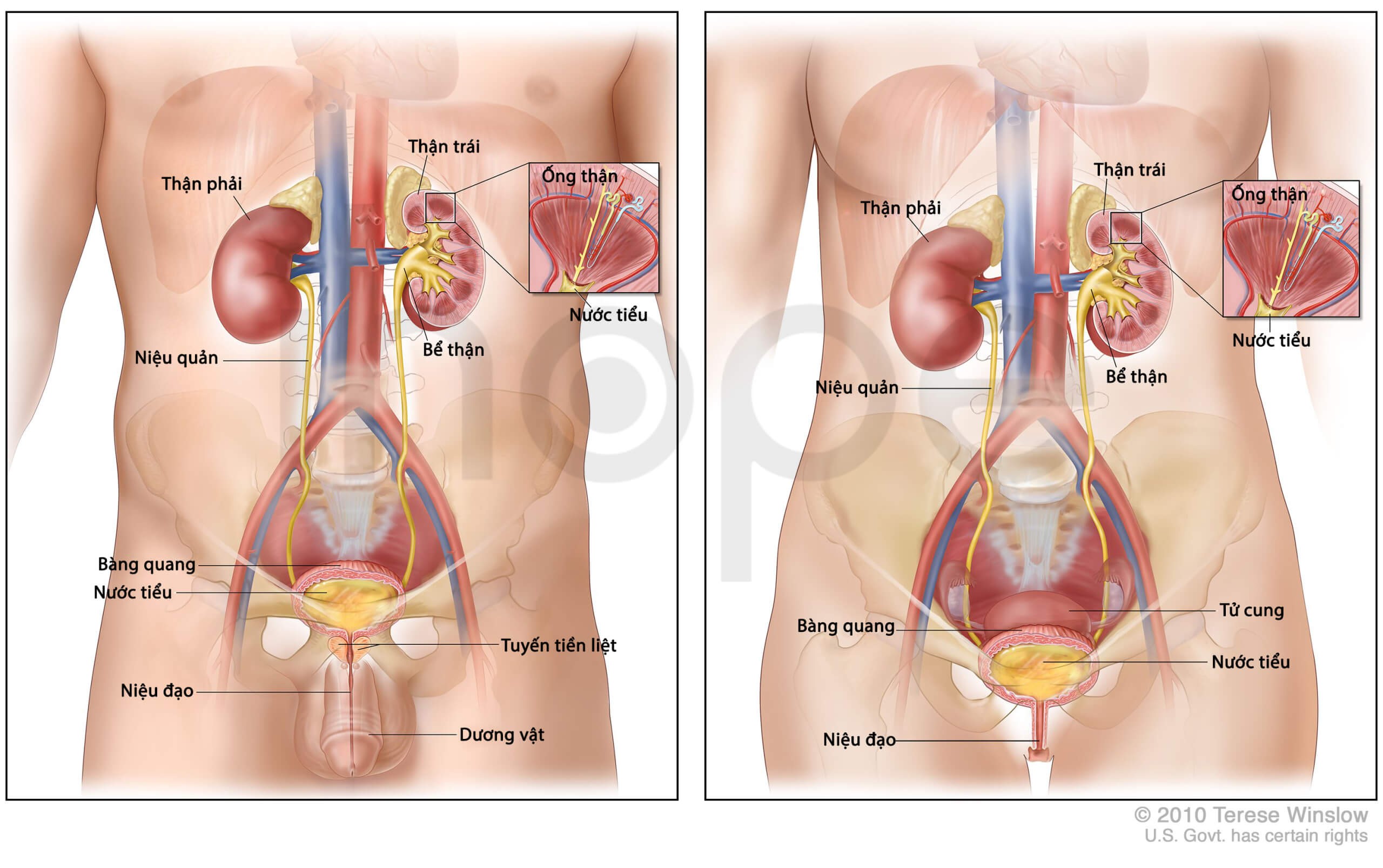
Nguồn: U.S. National Library of Medicine
Bên cạnh đó, một số người bệnh còn xuất hiện những triệu chứng sau:
- Hàm lượng axit uric trong máu cao do thận mất khả năng bài tiết axit này
- Hàm lượng bilirubin trong máu tăng gây vàng da và lòng trắng mắt
- Hàm lượng enzyme creatine kinase trong máu cao là dấu hiệu phổ biến của bệnh về cơ
Dạng nghiêm trọng
Bệnh dự trữ glycogen loại VII dạng nghiêm trọng thường khởi phát trong độ tuổi sơ sinh. Trẻ có dấu hiệu giảm trương lực cơ và tiến triển thành bệnh yếu cơ sau một thời gian. Ngoài ra, bệnh nhân còn có triệu chứng khó thở, tim yếu và các bệnh liên quan đến cơ tim. Trẻ bệnh thường không sống quá một năm sau khi sinh.
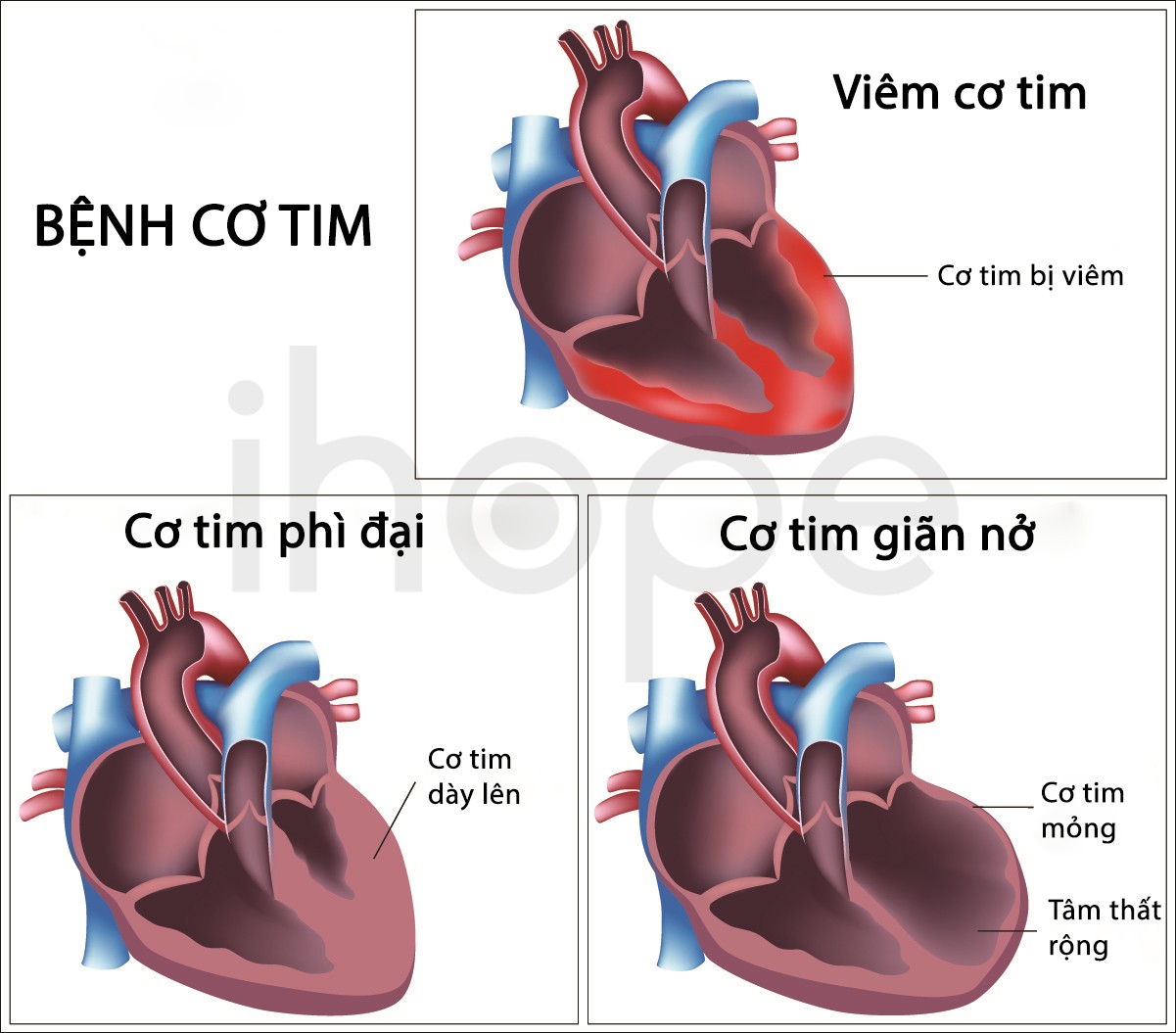
Nguồn: Alila Medical Media/Shutterstock.com
Dạng khởi phát muộn
Người bệnh dự trữ glycogen dạng khởi phát muộn thường xuất hiện triệu chứng yếu cơ. Bệnh khởi phát trong độ tuổi trưởng thành. Trong một số ít trường hợp, bệnh nhân gặp khó khăn khi vận động từ giai đoạn trẻ nhỏ. Bệnh chủ yếu ảnh hưởng đến cơ xung quanh vùng trung tâm của cơ thể.
Dạng tan máu
Dấu hiệu đặc trưng của bệnh dự trữ glycogen loại VII dạng tan máu là bệnh thiếu máu tán huyết. Tế bào hồng cầu trong cơ thể bệnh nhân vỡ sớm hơn so với bình thường nên hiện tượng thiếu máu xảy ra. Người bệnh không xuất hiện triệu chứng liên quan đến cơ.
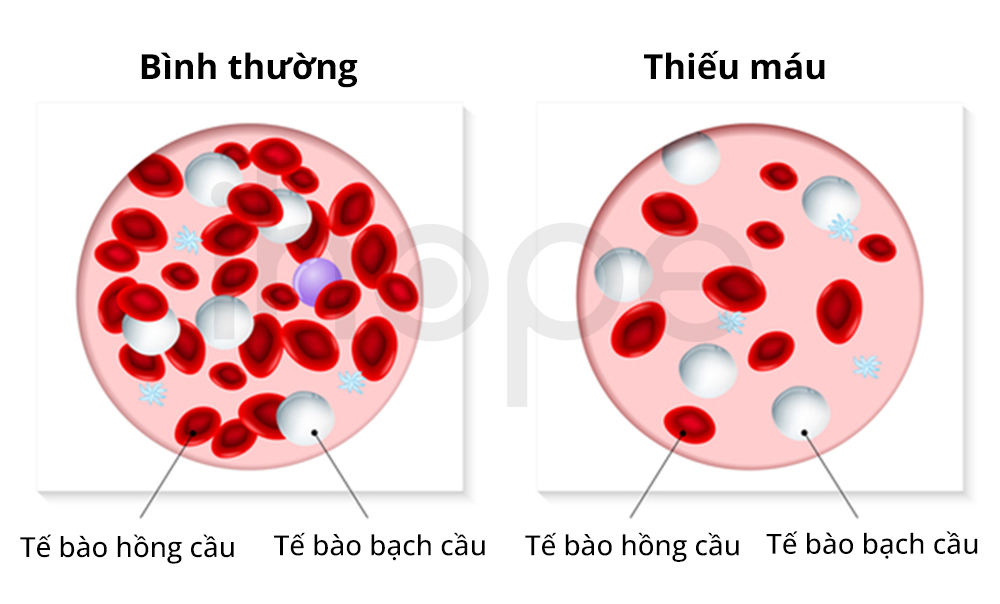
Nguồn: U.S. National Library of Medicine
Độ phổ biến
Bệnh dự trữ glycogen loại VII rất hiếm gặp. Hơn 100 trường hợp mắc bệnh đã được ghi nhận trên toàn thế giới.
Nguyên nhân
Đột biến gen PFKM gây ra bệnh dự trữ glycogen loại VII. Gen PFKM cung cấp hướng dẫn tạo ra tiểu đơn vị PFKM của enzyme phosphofructokinase. Enzyme này tham gia vào quá trình phân giải glycogen. Enzyme phosphofructokinase hiện diện trong nhiều loại mô, nó được cấu tạo từ bốn tiểu đơn vị. Các tiểu đơn vị khác nhau có mặt trong các loại mô khác nhau. Trong đó, tiểu đơn vị PFKM thường tồn tại trong cơ xương.

Nguồn: U.S National Library of Medicine
Trong cơ xương, nguồn năng lượng chính của tế bào thường dự trữ dưới dạng glycogen. Khi cơ thể cần duy trì hàm lượng glucose trong máu ổn định giữa các bữa ăn hoặc tạo năng lượng cho cơ thể vận động, glycogen sẽ nhanh chóng phân giải thành glucose. Phosphofructokinase tham gia vào nhiều quá trình phân giải glycogen để cung cấp năng lượng cho tế bào cơ.
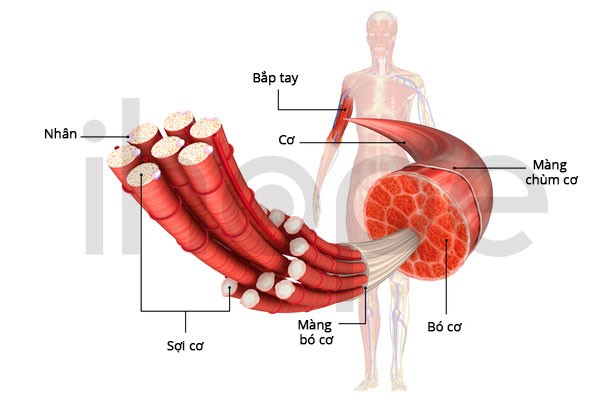
Nguồn: U.S. National Library of Medicine
Đột biến gen PFKM tạo ra các tiểu phần PFKM suy giảm hoặc không có chức năng khiến enzyme phosphofructokinase trong cơ xương mất chức năng nên không thể phân giải hoàn toàn glycogen. Cơ xương không nhận năng lượng từ glycogen sẽ trở nên yếu và chuột rút sau mỗi lần vận động hay tập thể dục với cường độ vừa phải. Trong một số trường hợp, cơ bắt đầu phân cắt. Đối với các mô khác trong cơ thể, các tiểu phần khác của enzyme phosphofructokinase có khả năng thay thế tiểu phần PFKM và duy trì hoạt động bình thường của enzyme. Do đó, đột biến gen PFKM không tác động lên những mô khác trong cơ thể.
Trong một số ít trường hợp, người bệnh xuất hiện triệu chứng nghiêm trọng hơn bình thường.
Chẩn đoán
Bác sĩ chẩn đoán bệnh dự trữ glycogen loại VII dựa trên hàm lượng enzyme phosphofructokinase bằng cách sinh thiết cơ hoặc kiểm tra tế bào hồng cầu.
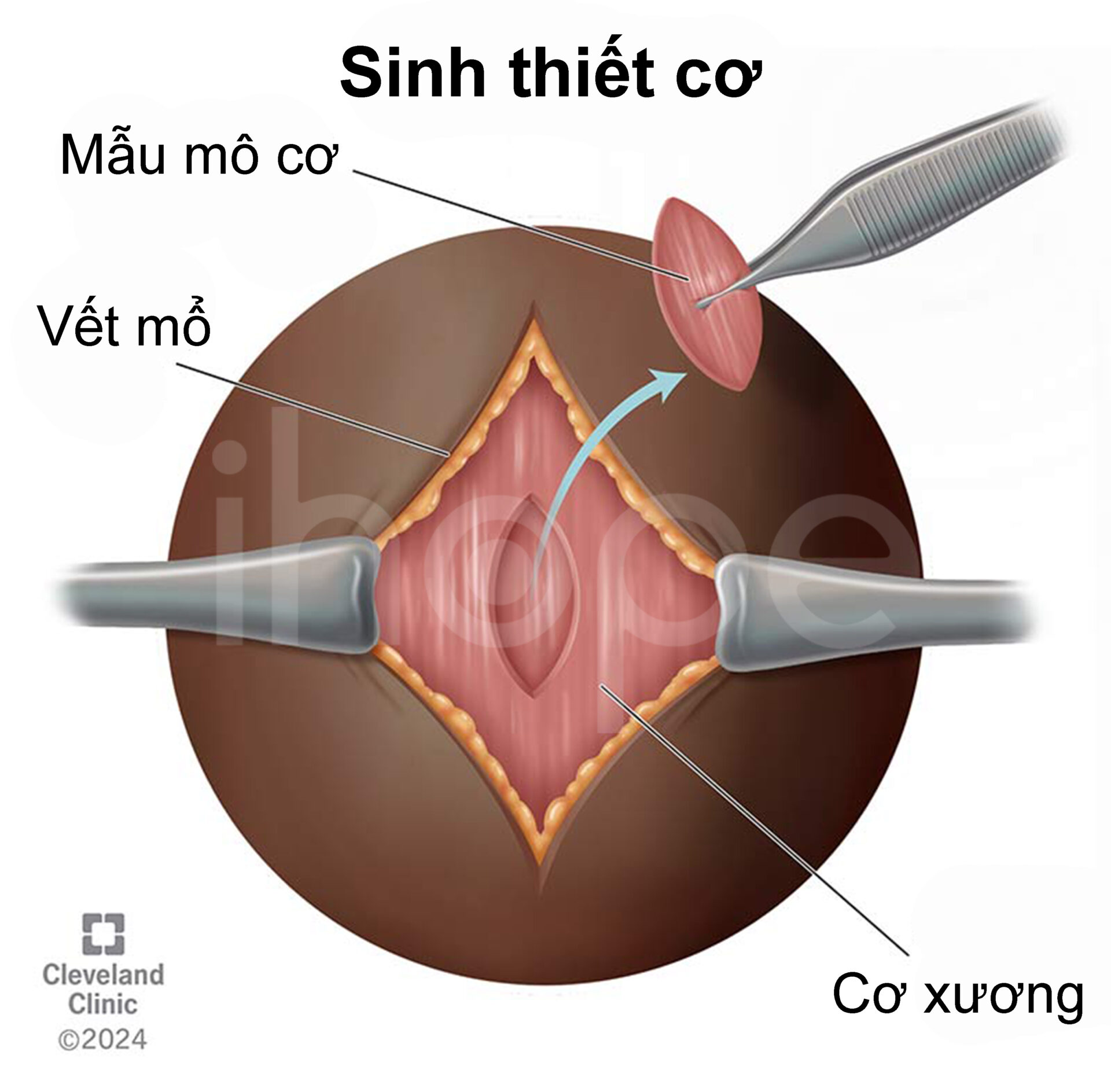
Nguồn: Cleveland Clinic
Ngoài ra, bác sĩ có thể chỉ định thực hiện một số xét nghiệm nhằm kiểm tra hàm lượng của protein creatinine kinase, lactate dehydrogenase và aspartate transaminase.
Mặt khác, người bệnh còn thực hiện sinh thiết cơ hoặc xét nghiệm máu sau khi vận động cẳng tay nhằm kiểm tra hàm lượng ammonia cao và lactate thấp. Khi cần thiết, bệnh nhân có thể xét nghiệm di truyền để xác nhận chẩn đoán.
Điều trị
Hiện nay, chưa có liệu pháp điều trị hoàn toàn bệnh dự trữ glycogen loại VII. Các phương pháp chủ yếu làm giảm triệu chứng bệnh và cải thiện chất lượng cuộc sống người bệnh.
Bệnh nhân nên áp dụng các phương pháp sau để kiểm soát triệu chứng của bệnh:
- Hạn chế vận động mạnh nhằm ngăn chặn đau cơ và chuột rút
- Giảm carbohydrate trong khẩu phần ăn để hạn chế tình trạng không dụng nạp sau khi vận động
- Bổ sung nhiều đạm vào chế độ ăn giúp giảm các triệu chứng của bệnh
Dạng di truyền
Bệnh dự trữ glycogen loại VII di truyền theo kiểu lặn trên nhiễm sắc thể thường, do đó cả hai bản sao của gen trong mỗi tế bào đều phải mang đột biến. Cha mẹ của một cá nhân mắc bệnh lặn trên nhiễm sắc thể thường đều mang một bản sao của gen đột biến, nhưng họ thường không biểu hiện dấu hiệu và triệu chứng của bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Bệnh dự trữ glycogen loại VII di truyền lặn do đột biến gen PFKM, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kì.
Các tên gọi khác
- Glycogenosis 7
- GSD VII
- GSD7
- Muscle phosphofructokinase deficiency
- PFKM deficiency
- Phosphofructokinase deficiency
- Tarui disease
References
- Genetic Testing Information. Glycogen storage disease, type VII. Retrieved July 25, 2024 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0017926/
- Genetic and Rare Diseases Information Center. Glycogen storage disease due to muscle phosphofructokinase deficiency. Retrieved July 25, 2024 from https://rarediseases.info.nih.gov/diseases/5686/index
- Catalog of Genes and Diseases from OMIM. GLYCOGEN STORAGE DISEASE VII; GSD7. Retrieved July 25, 2024 from https://omim.org/entry/232800
- U.S. National Library of Medicine. Glycogen storage disease type VII. Retrieved July 25, 2024 from https://medlineplus.gov/genetics/condition/glycogen-storage-disease-type-vii/
- Frontiers. Beneficial Effects of Ketogenic Diet on Phosphofructokinase Deficiency (Glycogen Storage Disease Type VII). Retrieved July 25, 2024 from https://www.frontiersin.org/journals/neurology/articles/10.3389/fneur.2020.00057/full
- MalaCards. Glycogen Storage Disease Vii (GSD7). Retrieved July 25, 2024 from https://www.malacards.org/card/glycogen_storage_disease_vii
- National Organization for Rare Disorders. Glycogen Storage Disease Type 7. Retrieved July 25, 2024 from https://rarediseases.org/rare-diseases/glycogen-storage-disease-type-vii/
- Orphanet. Glycogen storage disease due to muscle phosphofructokinase deficiency. Retrieved July 25, 2024 from https://www.orpha.net/en/disease/detail/371





