Bệnh lỗ não gia đình (familial porencephaly) là bệnh di truyền hiếm gặp, nguyên nhân bắt nguồn từ đột biến gen COL4A1 và gen COL4A2 trên nhiễm sắc thể 13. Một số biểu hiện đặc trưng của bệnh bao gồm nang chứa dịch trong não, não trắng và liệt nửa người.
Biểu hiện lâm sàng
Mức độ nghiêm trọng của các triệu chứng khác nhau giữa mỗi bệnh nhân. Bệnh có thể khởi phát trong giai đoạn thai kì hoặc sau khi sinh. Trong một số trường hợp, người bệnh chỉ biểu hiện triệu chứng khi trưởng thành.
Bệnh về não-thần kinh
Trẻ mắc bệnh có cấu trúc mạch máu dễ vỡ, dẫn đến xuất huyết và nang chứa dịch trong não. Những nang này thường xuất hiện tại một bên não, chúng có thể nối với vùng não thất hoặc khoang dưới nhện.
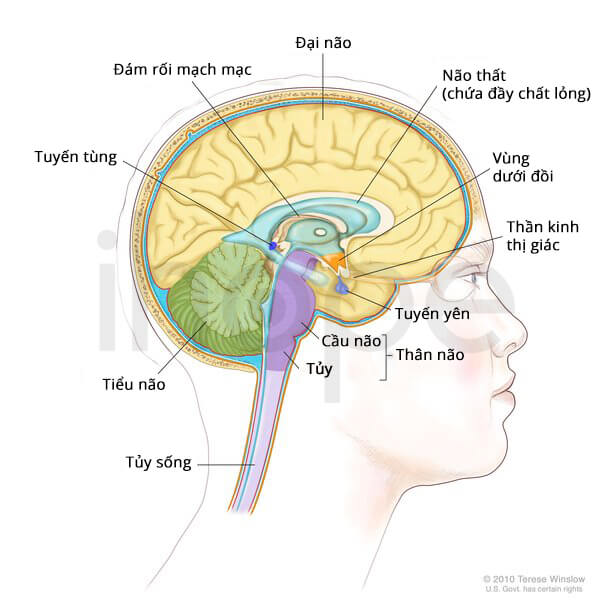
Nguồn: Terese Winslow
Ngoài ra, bệnh có thể gây ra một số bất thường về não bộ và hệ thần kinh như:
- Đột quỵ do xuất huyết não
- U nang não
- Não trắng
- Não úng thủy
- Co giật
- Liệt nửa người
- Đau nửa đầu
- Vôi hóa não
- Thiểu năng trí tuệ
Bệnh về mắt
Bệnh nhi thường có nhiều vấn đề với mắt như:
- Giác mạc nhỏ
- Đục thủy tinh thể
- Động mạch võng mạc cong bất thường
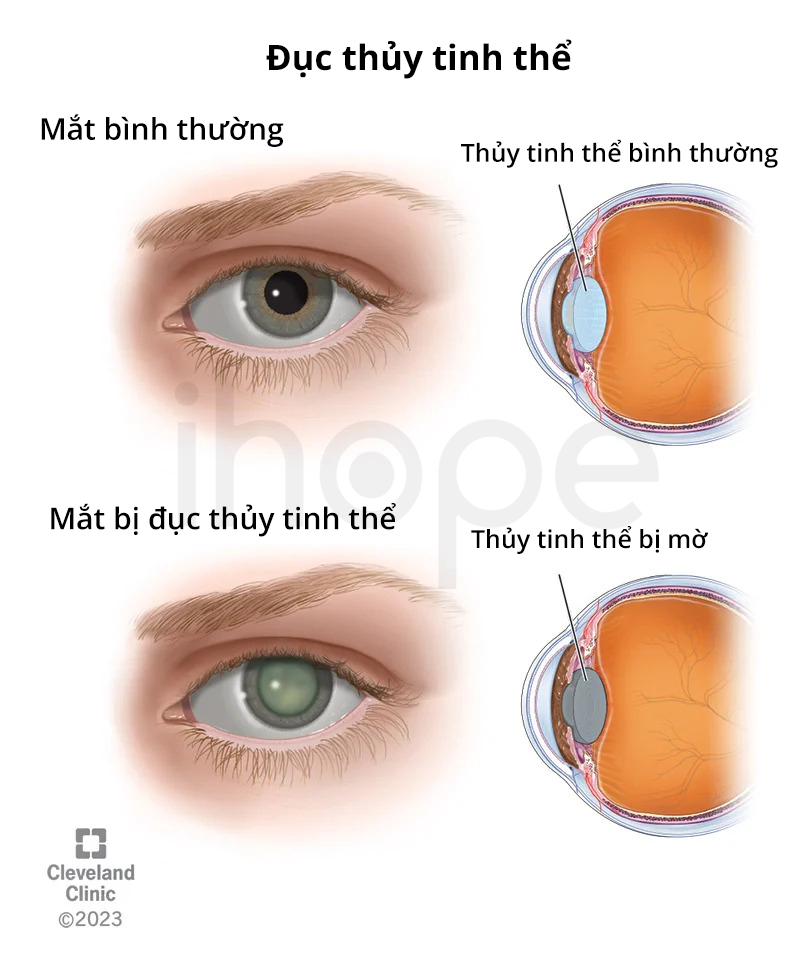
Nguồn: Cleveland Clinic
Triệu chứng khác
Ngoài ra, trẻ còn biểu hiện một số triệu chứng khác như:
- Đầu nhỏ
- U nang thận
- U nang gan
- Bất sản thận
- Chậm phát triển
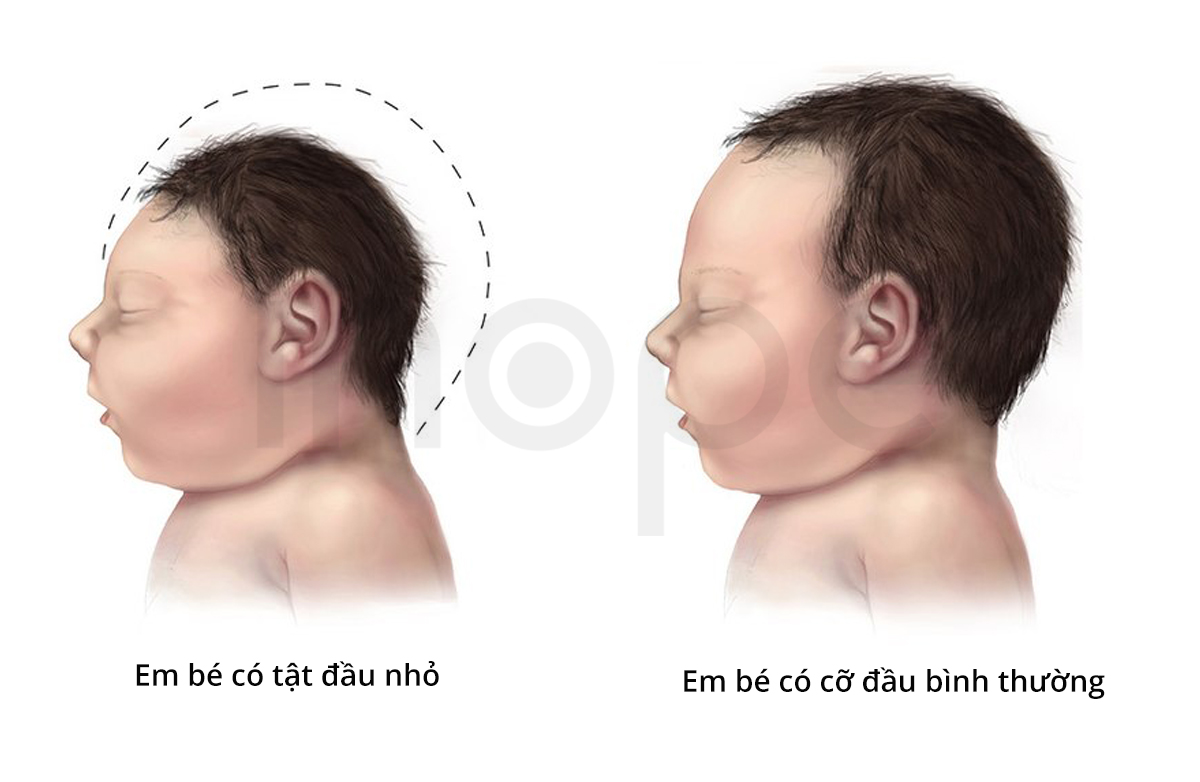
Nguồn: Centers for Disease Control and Prevention
Độ phổ biến
Bệnh lỗ não gia đình rất hiếm gặp nên tỉ lệ mắc bệnh chưa được xác định cụ thể. Hiện nay, người ta chỉ mới ghi nhận khoảng 8 gia đình mắc bệnh trên toàn thế giới.
Nguyên nhân
Bệnh lỗ não gia đình bắt nguồn từ đột biến một trong hai gen COL4A1 và COL4A2 trên nhiễm sắc thể 13. Gen COL4A1 và COL4A2 lần lượt cung cấp hướng dẫn tạo ra chuỗi alpha1(IV) và chuỗi alpha2(IV) của collagen loại IV—thành phần cấu tạo của lớp màng đáy bao quanh các mô và mạch máu trong cơ thể. Màng đáy nằm bên ngoài tế bào, chúng có dạng tấm mỏng để hỗ trợ duy trì cấu trúc tế bào cũng như phân tách các tế bào.
Đột biến gen COL4A1 hoặc COL4A2 thay đổi cấu trúc chuỗi alpha1(IV) hoặc alpha2(IV) nên các phân tử collagen loại IV không thể liên kết với nhau để hình thành mạng lưới protein tại màng đáy. Do đó, màng đáy mất tính ổn định nên không thể duy trì cấu trúc các mô. Đối với bệnh nhân mắc bệnh lỗ não gia đình, mạch máu não yếu đi dẫn đến vỡ mạch và nang chứa dịch trong não.
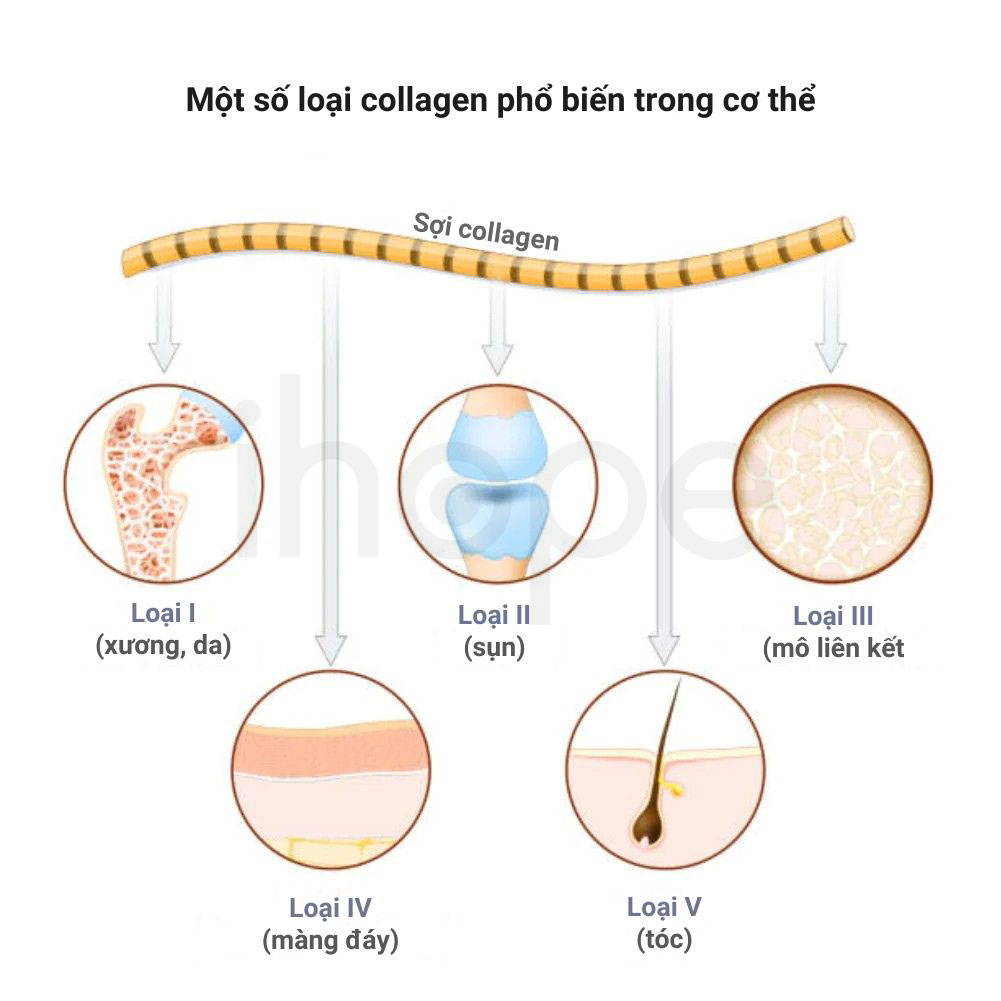
Nguồn: Immunescience
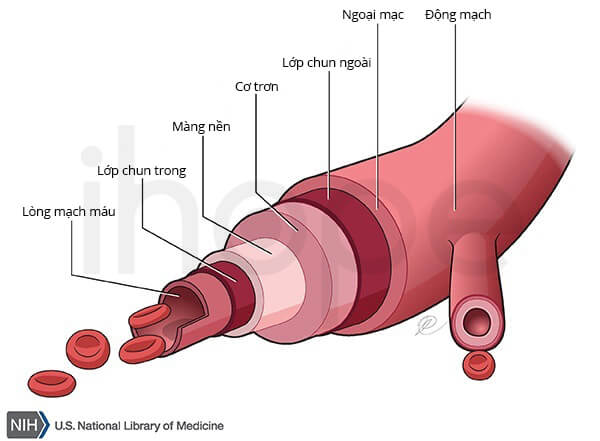
Nguồn: U.S. National Library of Medicine
Chẩn đoán
Bệnh lỗ não gia đình được chẩn đoán dựa trên biểu hiện lâm sàng, bệnh sử gia đình kết hợp với kết quả xét nghiệm hình ảnh và di truyền.
Chẩn đoán hình ảnh
Các phương pháp chẩn đoán hình ảnh não như MRI, chụp cắt lớp và siêu âm giúp bác sĩ phát hiện những đặc điểm bất thường trong não bộ của trẻ. Trong thai kì, bác sĩ có thể chẩn đoán bệnh thông qua siêu âm từ tuần thứ 30.
Xét nghiệm di truyền
Bác sĩ thực hiện các xét nghiệm di truyền nhằm phát hiện đột biến gen COL4A1 và COL4A2, qua đó chẩn đoán được xác nhận.
Điều trị
Hiện nay, chưa có phương pháp điều trị hoàn toàn bệnh lỗ não gia đình. Các liệu pháp chủ yếu nhằm giảm nhẹ triệu chứng và cải thiện chất lượng đời sống người bệnh.
Một số liệu pháp hỗ trợ bao gồm:
- Phẫu thuật điều trị đục thủy tinh thể
- Theo dõi mạch máu
- Sử dụng thuốc chống co giật
- Hạn chế sử dụng thuốc chống đông máu
- Trị liệu ngôn ngữ, hành vi
- Vật lí trị liệu
Dạng di truyền
Bệnh lỗ não gia đình di truyền theo kiểu trội trên nhiễm sắc thể thường. Do đó, một bản sao của gen đột biến trong mỗi tế bào đủ để gây ra bệnh. Người bệnh thường được di truyền từ cha hoặc mẹ mắc bệnh. Một số ít trường hợp do đột biến mới (de novo) xuất hiện trong quá trình hình thành tế bào sinh sản (trứng, tinh trùng) hoặc quá trình phát triển phôi sớm.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Bệnh lỗ não gia đình di truyền trội trên nhiễm sắc thể thường, nếu cha hoặc mẹ mắc bệnh, con sinh ra sẽ có 50% khả năng di truyền bệnh. Do đó, để bảo đảm 100% khả năng con không bị bệnh, cha mẹ có thể chọn phương pháp thụ tinh nhân tạo IVF và sàng lọc phôi PGS/PGD. Các thành viên trong gia đình nên khám sức khỏe và tầm soát bệnh định kì nếu có thành viên mắc bệnh. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh.
Các tên gọi khác
- Autosomal dominant porencephaly type 1
- Infantile hemiplegia with porencephaly
- Porencephaly type 1
- Hereditary porencephaly
- Familial porencephalic cyst
- Familial porencephalic white matter disease
References
- National Library of Medicine. Familial porencephaly. Retrieved July 17, 2025 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C1867983/
- Genetic and Rare Diseases Information Center. Familial porencephaly. Retrieved July 17, 2025 from https://rarediseases.info.nih.gov/diseases/2258/index
- OMIM. BRAIN SMALL VESSEL DISEASE 1 WITH OR WITHOUT OCULAR ANOMALIES; BSVD1. Retrieved July 17, 2025 from https://omim.org/entry/175780
- MedlinePlus. Familial porencephaly. Retrieved July 17, 2025 from https://medlineplus.gov/genetics/condition/familial-porencephaly/
- GeneReviews. COL4A1-Related Disorders. Retrieved July 17, 2025 from https://www.ncbi.nlm.nih.gov/books/NBK7046/
- MedGen. Familial porencephaly. Retrieved July 17, 2025 from https://www.ncbi.nlm.nih.gov/medgen/401353
- Orphanet. Porencephaly. Retrieved July 17, 2025 from https://www.orpha.net/en/disease/detail/2940
- Springer Nature. COL4A2 mutation associated with familial porencephaly and small-vessel disease. Retrieved July 17, 2025 from https://doi.org/10.1038/ejhg.2012.20