
Thoái hóa tiểu não loại 6 (spinocerebellar ataxia type 6) là bệnh di truyền khiến người bệnh khó phối hợp cử động và giữ thăng bằng. Khả năng vận động của người bệnh giảm dần theo thời gian.
Biểu hiện lâm sàng
Biểu hiện của thoái hóa tiểu não loại 6 thường bắt đầu tại độ tuổi 40 hoặc 50. Tuy nhiên, bệnh có thể khởi phát bất cứ lúc nào từ thời thơ ấu đến cuối tuổi trưởng thành. Người bệnh cần hỗ trợ đi bộ hoặc di chuyển khi về già.
Các dấu hiệu và triệu chứng sớm của thoái hóa tiểu não loại 6 bao gồm:
- Nói chuyện khó khăn
- Chuyển động mắt không tự chủ (rung giật nhãn cầu)
- Song thị
Theo thời gian, người bệnh có thể mất khả năng phối hợp tay, run và căng cơ không kiểm soát.
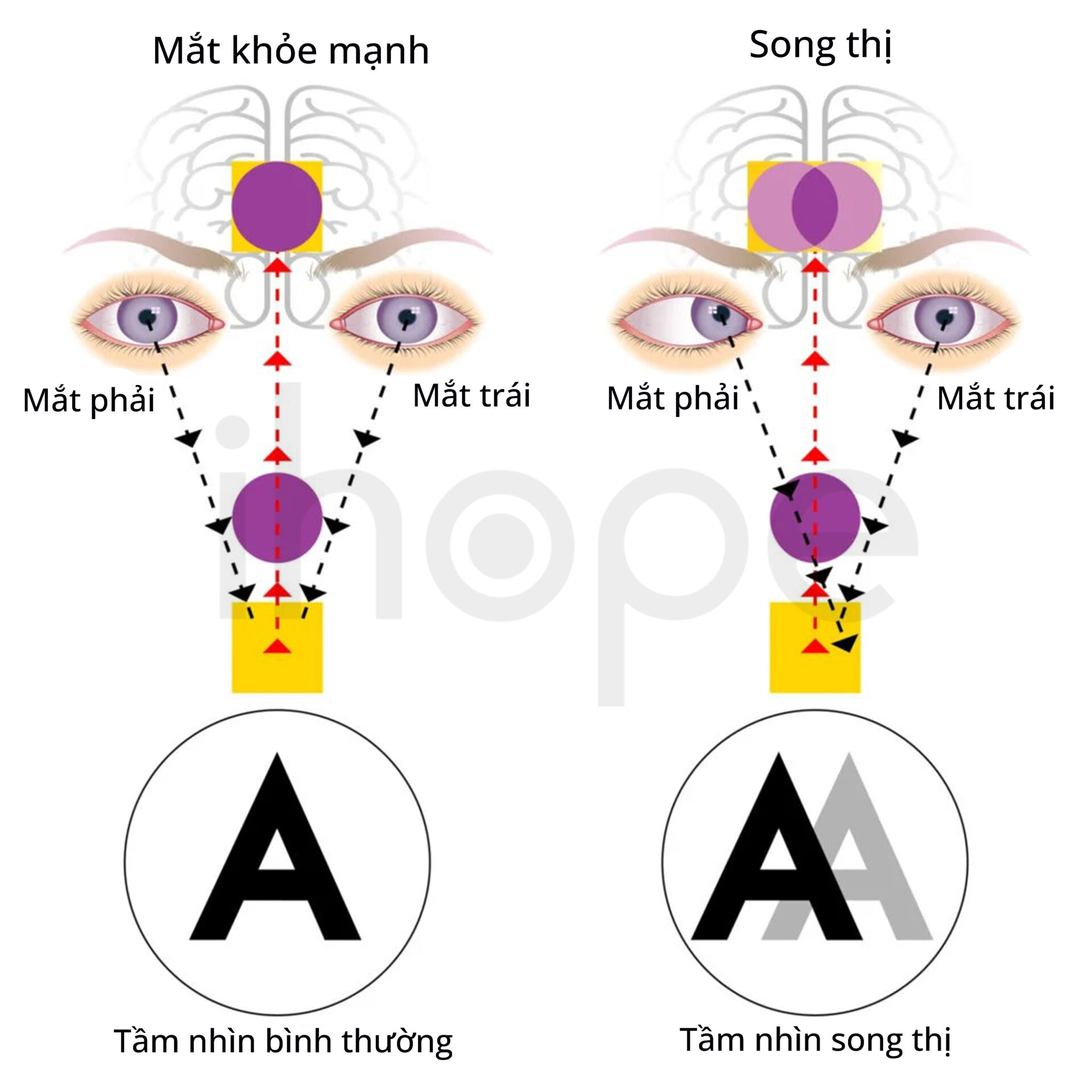
Ảnh: Tầm nhìn đôi (song thị)
Nguồn: Vision Center
Độ phổ biến
Ước tính tỉ lệ mắc bệnh thoái hóa tiểu não loại 6 trên toàn thế giới chưa đến 1/100.000 người.
Nguyên nhân
Đột biến gen CACNA1A gây ra thoái hóa tiểu não loại 6. Gen này cung cấp hướng dẫn tạo ra tiểu đơn vị alpha-1 của kênh canxi CaV2.1.
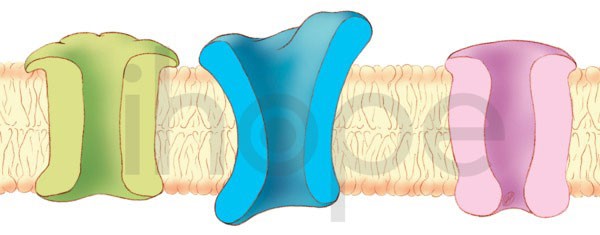
Nguồn: U.S. National Library of Medicine
Kênh CaV2.1 có chức năng rất quan trọng đối với hoạt động giao tiếp giữa các tế bào thần kinh trong não. Cụ thể, kênh này vận chuyển ion canxi tích điện dương qua màng tế bào. Quá trình di chuyển ion canxi giúp tế bào thần kinh trao đổi tín hiệu với những tế bào thần kinh khác trong não cũng như các bộ phận khác của hệ thần kinh .
Nguồn: Eunice Kennedy Shriver National Institute of Child Health and Human Development
Đột biến gen CACNA1A gây ra thoái hóa tiểu não loại 6 liên quan đến lặp lại ba nucleotide CAG. Đoạn lặp này bao gồm ba nucleotide cytosine, adenine và guanine lặp lại nhiều lần. Thông thường, đoạn CAG lặp lại từ 4–18 lần trên gen. Tuy nhiên, đoạn CAG lặp lại từ 20–33 lần đối với người bệnh. Số lượng đoạn lặp liên quan đến độ tuổi khởi phát bệnh. Người bệnh mang 20 đoạn lặp CAG có xu hướng khởi phát bệnh khi họ từ 65 tuổi trở lên. Trong khi đó, người bệnh có số đoạn lặp nhiều hơn thường bắt đầu biểu hiện bệnh từ 40–65 tuổi.
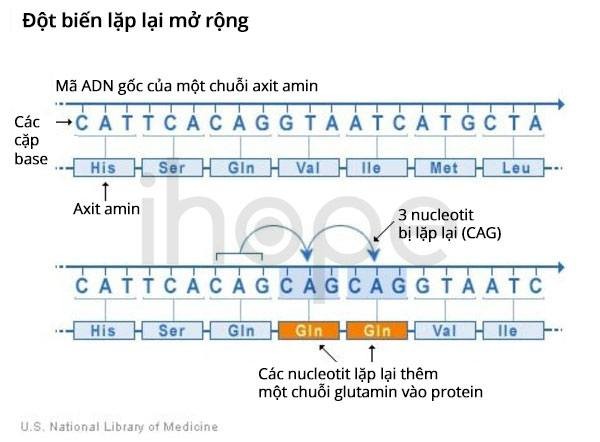
Nguồn: U.S. National Library of Medicine
Số lượng đoạn lặp ba nucleotide CAG tăng lên dẫn đến sản xuất ra tiểu đơn vị alpha-1 dài bất thường, do đó ảnh hưởng đến cấu trúc và chức năng của kênh CaV2.1. Thông thường, kênh CaV2.1 nằm bên trong màng tế bào. Tuy nhiên, kênh CaV2.1 bất thường hiện diện trong màng tế bào cũng như tế bào chất. Chúng tụ lại với nhau tạo thành các cục trong tế bào chất. Người ta chưa rõ các cục này ảnh hưởng như thế nào đến hoạt động của tế bào. Mặt khác, thiếu kênh CaV2.1 trong màng tế bào làm suy yếu hoạt động giao tiếp giữa các tế bào thần kinh trong não, dẫn đến chết tế bào. Thiếu kênh CaV2.1 ảnh hưởng nghiêm trọng đến tế bào trong tiểu não có chức năng điều phối chuyển động. Do đó, người bệnh mất dần khả năng vận động theo thời gian.
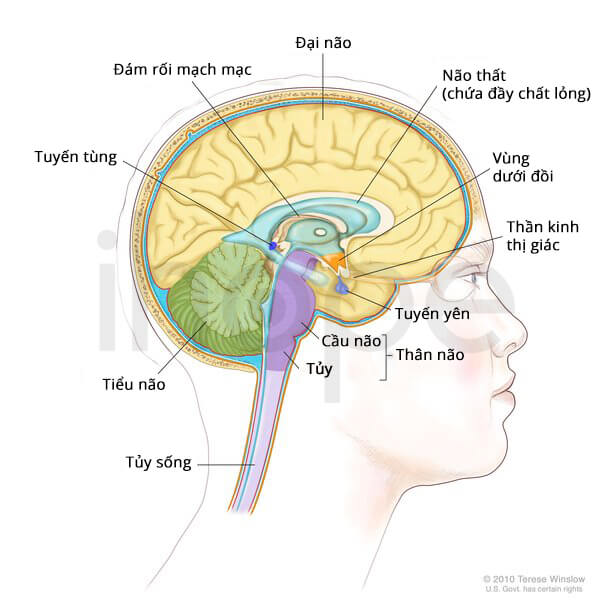
Nguồn: Terese Winslow
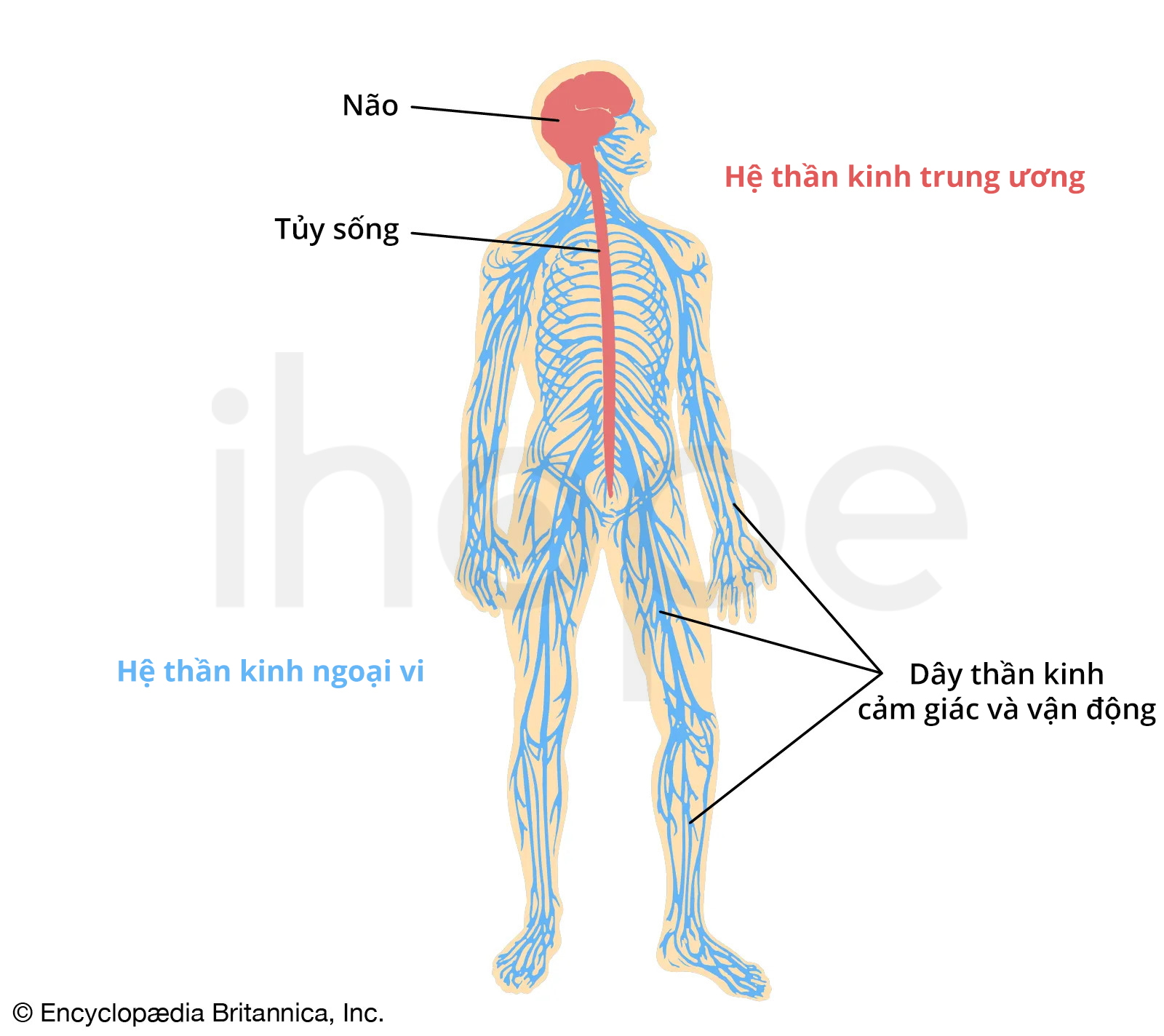
Ảnh: Hệ thần kinh người
Nguồn: Encyclopædia Britannica, Inc
Chẩn đoán
Thoái hóa tiểu não loại 6 được chẩn đoán dựa trên đánh giá biểu hiện lâm sàng, xem xét bệnh sử cá nhân, gia đình kết hợp với thực hiện xét nghiệm để phát hiện bất thường trong não.
Một số xét nghiệm chụp ảnh não bao gồm:
Ngoài ra, người bệnh có thể thực hiện xét nghiệm di truyền nhằm xác định đột biến gen CACNA1A, qua đó kết quả chẩn đoán được xác nhận.
Điều trị
Hiện nay, chưa có phương pháp điều trị hoàn toàn thoái hóa tiểu não loại 6. Các liệu pháp chủ yếu nhằm giảm nhẹ triệu chứng và cải thiện chất lượng đời sống người bệnh.
Các biện pháp điều trị thoái hóa tiểu não loại 6 phổ biến gồm:
- Sử dụng thiết bị hỗ trợ di chuyển như nạng, xe lăn
- Vật lí trị liệu để tăng cường cơ bắp, cải thiện dáng đi và khả năng giữ thăng bằng
- Sử dụng thuốc giúp giảm run rẩy, cứng cơ và co thắt cơ
Dạng di truyền
Thoái hóa tiểu não loại 6 di truyền theo kiểu trội trên nhiễm sắc thể thường. Do đó, một bản sao của gen trong mỗi tế bào đủ để gây ra bệnh. Người bệnh thường được di truyền từ cha hoặc mẹ mắc bệnh. Một số ít trường hợp bắt nguồn từ đột biến mới (denovo) trong quá trình hình thành tế bào sinh sản (trứng, tinh trùng) hoặc quá trình phát triển phôi sớm.

Nguồn: U.S. National Library of Medicine
Khi đột biến gen CACNA1A được truyền từ thế hệ này sang thế hệ khác, độ dài của đoạn lặp lại ba nucleotide CAG thường tăng nhẹ. Số lượng đoạn lặp lại tăng liên quan đến độ tuổi khởi phát, do đó các dấu hiệu và triệu chứng bệnh khởi phát sớm hơn.
Phòng ngừa
Thoái hóa tiểu não loại 6 di truyền trội trên nhiễm sắc thể thường, nếu cha hoặc mẹ mắc bệnh, con sinh ra sẽ có 50% khả năng di truyền bệnh. Do đó, để bảo đảm 100% khả năng con không bị bệnh, cha mẹ có thể chọn phương pháp thụ tinh nhân tạo IVF và sàng lọc phôi PGS/PGD. Các thành viên trong gia đình nên khám sức khỏe và tầm soát bệnh định kì nếu có thành viên mắc bệnh. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh.
Các tên gọi khác
- SCA6
- Type 6 spinocerebellar ataxia
References
- Genetic Testing Information. Spinocerebellar ataxia type 6. Retrieved January 13, 2025 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0752124/
- Genetic and Rare Diseases Information Center. Spinocerebellar ataxia type 6. Retrieved January 13, 2025 from https://rarediseases.info.nih.gov/diseases/10351/index
- Catalog of Genes and Diseases from OMIM. SPINOCEREBELLAR ATAXIA 6; SCA6. Retrieved January 13, 2025 from https://omim.org/entry/183086
- U.S National Library of Medicine. Spinocerebellar ataxia type 6. Retrieved January 13, 2025 from https://medlineplus.gov/genetics/condition/spinocerebellar-ataxia-type-6/
- Frontiers. Molecular mechanism of Spinocerebellar Ataxia type 6: glutamine repeat disorder, channelopathy and transcriptional dysregulation. The multifaceted aspects of a single mutation. Retrieved January 13, 2025 from https://www.frontiersin.org/journals/cellular-neuroscience/articles/10.3389/fncel.2015.00036/full
- MalaCards. Spinocerebellar Ataxia 6 (SCA6). Retrieved January 13, 2025 from https://www.malacards.org/card/spinocerebellar_ataxia_6
- National Institute of Health. Spinocerebellar Ataxia Type 6. Retrieved January 13, 2025 from https://www.ncbi.nlm.nih.gov/books/NBK1140/
- Orphanet. Spinocerebellar ataxia type 6. Retrieved January 13, 2025 from https://www.orpha.net/en/disease/detail/98758





