Hội chứng Cohen là bệnh di truyền ảnh hưởng đến nhiều bộ phận của cơ thể. Người bệnh thường bị yếu cơ, chậm phát triển cả về tinh thần và thể chất và khuôn mặt bất thường.
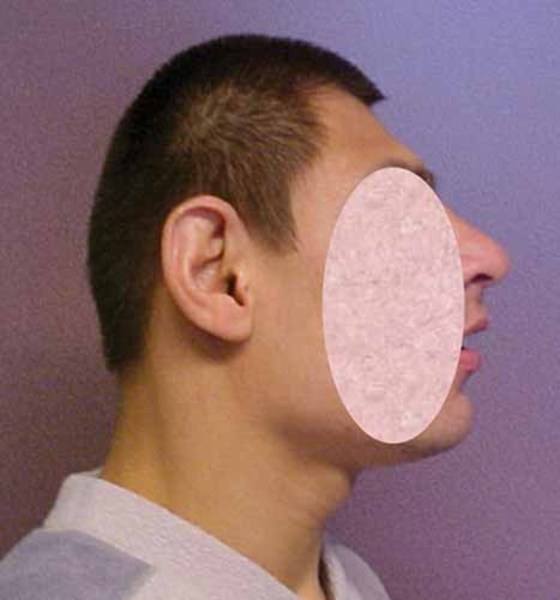
Ảnh: Dị tật đầu nhỏ
Nguồn: Elements of Morphology, National Human Genome Research Institute
Biểu hiện lâm sàng
Các dấu hiệu và triệu chứng của hội chứng Cohen có thể khác nhau ở mỗi bệnh nhân. Trẻ sơ sinh mắc bệnh thường yếu cơ vài ngày đầu sau sinh. Một số trẻ có thể có tiếng khóc yếu ớt hoặc the thé, chậm phát triển, kích thước đầu nhỏ (tật đầu nhỏ ).
Khi lớn hơn, chúng có biểu hiện chậm phát triển như không thể tự ngồi, lăn lộn, chậm biết đi (thường 2-5 tuổi mới biết đi). Mức độ biểu hiện bệnh khác nhau ngay cả giữa các thành viên trong cùng một gia đình.
Các đặc điểm khác hay gặp bao gồm cận thị tiến triển nặng theo thời gian, thoái hóa mô nhạy cảm với ánh sáng ở phía sau của mắt (loạn dưỡng võng mạc), khớp lỏng lẻo. Khuôn mặt dị biệt bao gồm nhiều lông và lông mày dày, lông mi dài, đôi mắt có hình dạng bất thường (xếch xuống), đầu mũi to, vùng nhân trung phẳng, răng cửa nổi rõ làm người bệnh khó khép kín miệng.
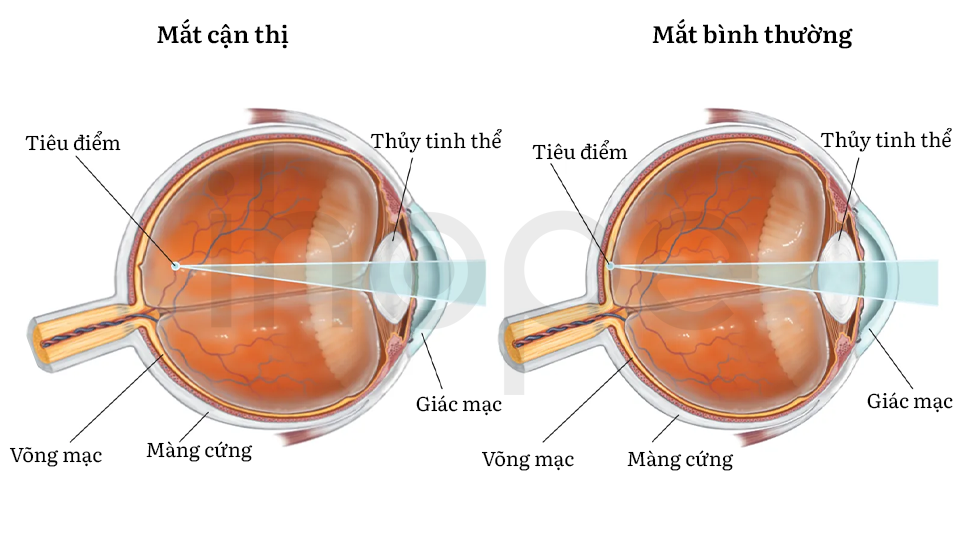
Nguồn: Encyclopaedia Britannica
Đôi khi, người bệnh bị giảm bạch cầu trung tính, béo phì thường phát triển ở trẻ em hoặc thanh thiếu niên, tầm vóc thấp. Mô mỡ tích tụ xung quanh thân nhưng cánh tay và chân ốm yếu.
Biến chứng
Người mắc hội chứng Cohen có nguy cơ cao phát triển các bệnh tự miễn dịch, đặc biệt tiểu đường, bệnh tuyến giáp và bệnh Celiac. Bệnh tự miễn xảy ra khi hệ miễn dịch tấn công nhầm các mô khỏe mạnh.
Độ phổ biến
Người ta vẫn chưa xác định chính xác tỷ lệ mắc bệnh của hội chứng Cohen, khoảng 1.000 trường hợp mắc bệnh trên toàn thế giới đã được ghi nhận. Bệnh ảnh hưởng đến nam và nữ với tỷ lệ ngang nhau. Tuy nhiên, các trường hợp mắc bệnh thường không được chẩn đoán hoặc chẩn đoán sai, gây khó khăn cho quá trình xác định tần suất mắc bệnh thực sự.
Nguyên nhân
Đột biến gen VPS13B (còn gọi là gen COH1) gây ra hội chứng Cohen. Gen VPS13B cung cấp hướng dẫn tạo ra một loại protein tham gia vào cấu trúc tế bào của bộ máy Golgi. Cụ thể hơn, protein VPS13B được biến đổi thông qua quá trình glycosyl hóa - quá trình gắn các phân tử đường vào protein. Protein VPS13B còn tham gia vào quá trình phân loại và vận chuyển protein bên trong tế bào. Ngoài ra, protein VPS13B còn liên quan đến quá trình tăng trưởng và phát triển bình thường của tế bào thần kinh và tế bào mỡ, nó còn có thể tham gia lưu trữ và phân phối chất béo trong cơ thể.
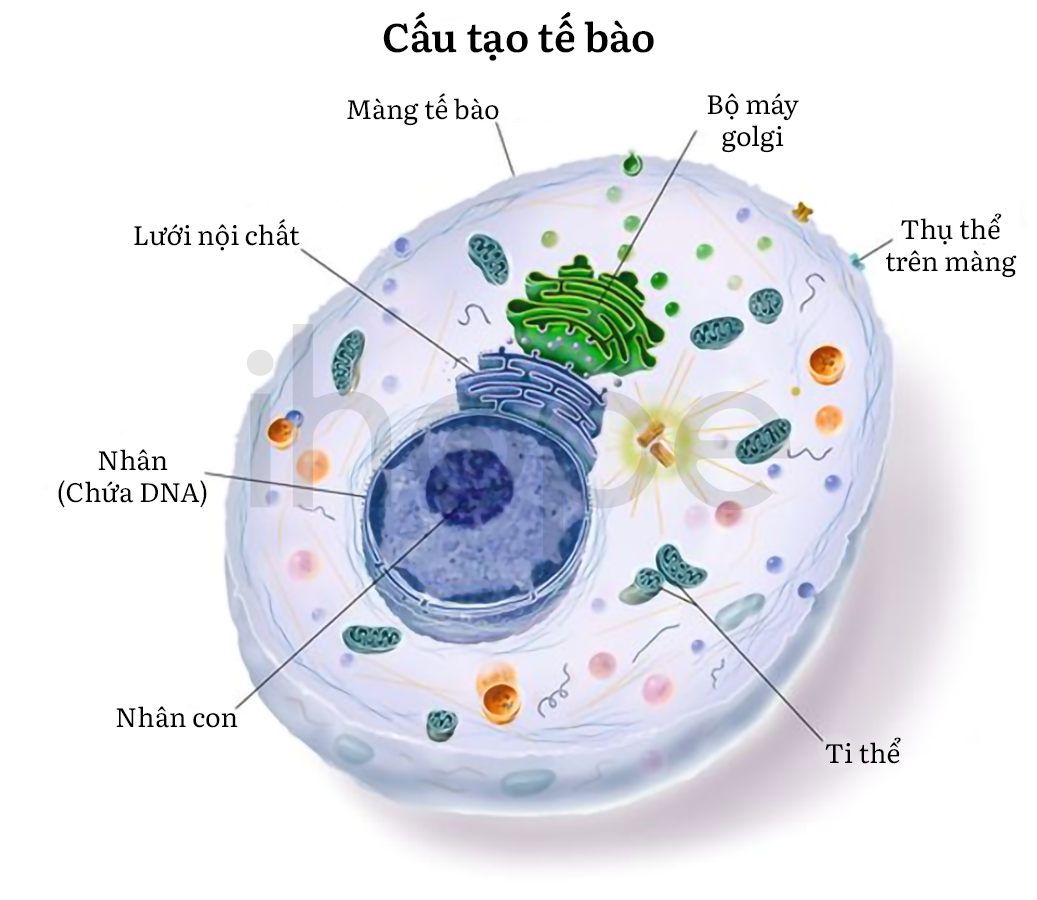
Nguồn: National Cancer Institue
Hầu hết các đột biến gen VPS13B ngăn cản quá trình sản xuất protein VPS13B, dẫn đến phá vỡ tổ chức của bộ máy Golgi và suy giảm quá trình glycosyl hóa. Tuy nhiên, người ta vẫn chưa hiểu rõ cơ chế ảnh hưởng khi protein VPS13B bị thiếu hoặc những thay đổi trong tế bào dẫn đến triệu chứng lâm sàng. Người ta cho rằng các vấn đề về phát triển tế bào thần kinh là nguyên nhân dẫn đến tật đầu nhỏ, thiểu năng trí tuệ, loạn dưỡng võng mạc và tích trữ chất béo bất thường có thể gây béo phì ở những người mắc hội chứng Cohen.
Chẩn đoán
Khám sức khỏe nhằm tìm các dấu hiệu của bệnh và tiền sử bệnh trong gia đình.
Xét nghiệm di truyền tìm đột biến gen VPS13B đối với trường hợp nghi ngờ mắc hội chứng Cohen.
Chấn đoán phân biệt
Hội chứng Prader-Willi là bệnh di truyền gây ra các biểu hiện đặc trưng trong thời kỳ sơ sinh bao gồm yếu cơ, khó ăn và chậm tăng cân. Trẻ thường chậm phát triển, tầm vóc thấp, bộ phận sinh dục nhỏ và thèm ăn quá mức dẫn đến béo phì. Người bị béo phì nặng có thể tăng nguy cơ suy tim, ngưng thở khi ngủ, tiểu đường, từ đó có thể gây ra các biến chứng đe dọa tính mạng. Người bệnh thường bị thiểu năng trí tuệ nhẹ đến trung bình. Bệnh xảy ra do đột biến một số gen trên nhiễm sắc thể số 15.
Hội chứng Angelman là bệnh thần kinh di truyền hiếm gặp, bệnh đặc trưng bởi biểu hiện chậm phát triển nghiêm trọng, thiểu năng trí tuệ, mất khả năng phối hợp các cử động, biểu hiện vui và cười vô cớ. Trẻ không nói được nhưng có thể học cách giao tiếp qua cử chỉ và hiểu các lệnh đơn giản. Các triệu chứng bổ sung có thể xảy ra trong một số trường hợp bao gồm co giật, khó ngủ và khó ăn hoặc khuôn mặt dị biệt. Hội chứng Angelman xảy ra do gen UBE3A bị mất hoặc biểu hiện bất thường.
Một số rối loạn di truyền khác có thể biểu hiện các dấu hiệu và triệu chứng tương tự như hội chứng Cohen bao gồm hội chứng Alstrom, hội chứng Cri-du-chat, hội chứng Williams, hội chứng Bardet-Biedl và suy giáp bẩm sinh.
Điều trị
Quá trình điều trị hội chứng Cohen tập trugng vào các triệu chứng cụ thể ở mỗi người bệnh.
Can thiệp sớm rất quan trọng giúp khắc phục tối đa những biến chứng của bệnh.
- Hầu hết trẻ mắc bệnh được áp dụng các liệu pháp vật lý, ngôn ngữ và hành vi
- Đeo mắt kính giúp tăng thị lực
- Nhiễm trùng tái phát có thể điều trị bằng kháng sinh
Trong một số trường hợp, giảm bạch cầu trung tính có thể điều trị bằng cách sử dụng các yếu tố kích thích tế bào bạch cầu hạt (G-CSF) kích thích tủy xương sản xuất bạch cầu trung tính và cải thiện khả năng tiêu diệt vi khuẩn của chúng.
Dạng di truyền
Bệnh di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, hai bản sao gen đột biến trong tế bào mới có khả năng gây ra bệnh. Người bệnh có bố mẹ mang gen bị bệnh nhưng không biểu hiện triệu chứng của bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Hiện không có biện pháp phòng ngừa hội chứng Cohen vì đây là bệnh di truyền. Các thành viên trong gia đình nên khám sức khỏe và tầm soát bệnh định kỳ nếu có thành viên mắc bệnh. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh.
Bệnh di truyền lặn đột biến gen VPS13B, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con.
Các tên gọi khác
- Hypotonia, obesity, and prominent incisors
- Norio syndrome
- Obesity-hypotonia syndrome
- Pepper syndrome
- Prominent incisors-obesity-hypotonia syndrome
References
- Genetic Testing Information. Cohen syndrome. Retrieved Aug 20, 2022 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0265223/
- Genetic and Rare Diseases Information Center. Cohen syndrome. Retrieved Aug 20, 2022 from https://rarediseases.info.nih.gov/diseases/6126/cohen-syndrome
- Catalog of Genes and Diseases from OMIM. COHEN SYNDROME; COH1 Retrieved Aug 20, 2022 from https://omim.org/entry/216550
- U.S National Library of Medicine. Cohen syndrome. Retrieved Aug 20, 2022 from https://medlineplus.gov/genetics/condition/cohen-syndrome
- National Organization for Rare Disorders. Cohen Syndrome. Retrieved Aug 20, 2022 from https://rarediseases.org/rare-diseases/cohen-syndrome/
- National Library of Medicine. Cohen Syndrome. Retrieved Aug 20, 2022 from https://www.ncbi.nlm.nih.gov/books/NBK1482/
- Mala Cards - Human Disease Database. Cohen Syndrome (COH1). Retrieved Aug 20, 2022 from https://www.malacards.org/card/cohen_syndrome
- Disease InfoSearch. Cohen Syndrome (COH1). Retrieved Aug 20, 2022 from https://www.diseaseinfosearch.org/Cohen+Syndrome/1715
- PubMed. Gut fermentation syndrome: A systematic review of case reports. Retrieved Aug 20, 2022 from https://pubmed.ncbi.nlm.nih.gov/33887125/