Hội chứng Knobloch (Knobloch syndrome) là bệnh di truyền hiếm gặp, người bệnh biểu hiện các triệu chứng nghiêm trọng về thị lực và khiếm khuyết hộp sọ.
Biểu hiện lâm sàng
Dấu hiệu đặc trưng của hội chứng Knobloch là cận thị nặng. Ngoài ra, bệnh còn gây ra những bất thường về mắt khác. Phần lớn người bệnh bị thoái hóa dịch kính võng mạc, trong đó thủy tinh thể và võng mạc bị phá vỡ. Thủy tinh thể giống như gelatin lấp đầy mắt và võng mạc, mô nhạy cảm với ánh sáng phía sau mắt. Thoái hóa dịch kính võng mạc thường dẫn đến bong võng mạc . Một số trường hợp gặp bất thường tại điểm vàng, khu vực trung tâm của võng mạc. Điểm vàng mang lại tầm nhìn trung tâm sắc nét, các tác vụ như đọc, lái xe, nhận diện khuôn mặt cần khả năng này. Do bất thường thủy tinh thể, võng mạc và điểm vàng, người mắc hội chứng Knobloch thường bị mù một hoặc cả hai mắt.
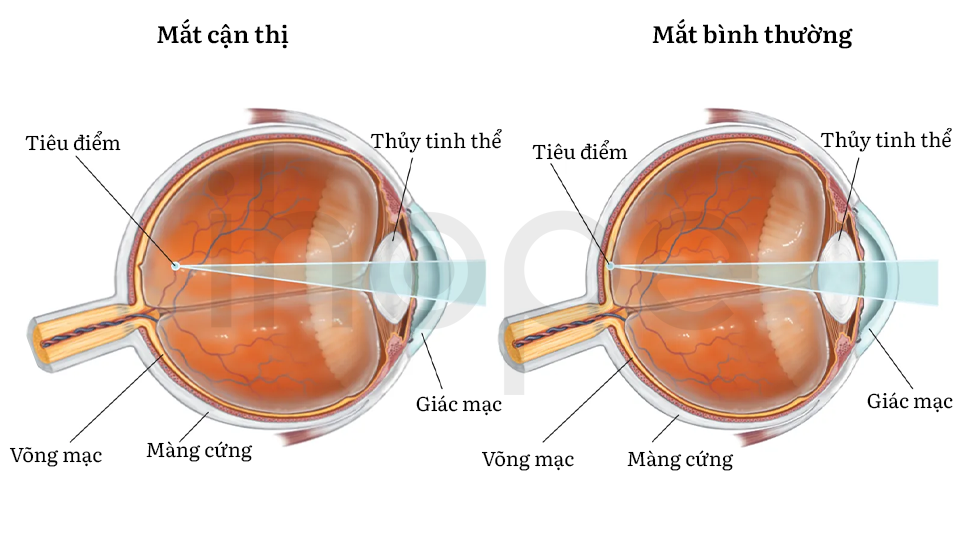
Ảnh: Cận thị
Nguồn: Encyclopaedia Britannica
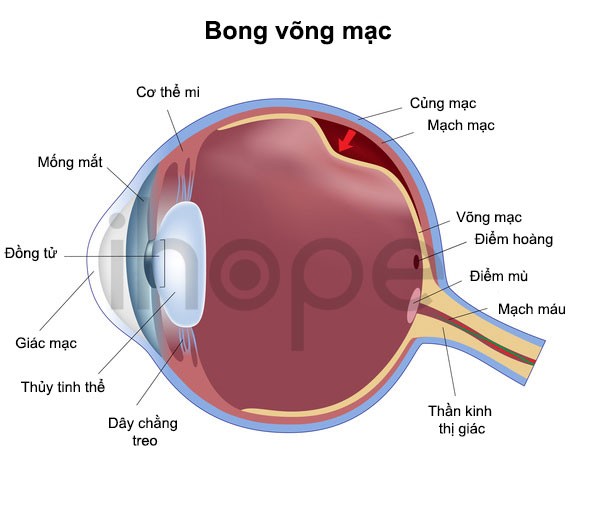
Ảnh: Bong võng mạc
Nguồn: U.S. National Library of Medicine
Một dấu hiệu đặc trưng khác của hội chứng Knobloch là thoát vị não chẩm , một phần nhô ra giống như túi phía sau đầu thông qua khiếm khuyết trong xương vùng đáy hộp sọ (xương chẩm). Đối với các bệnh lý khác, thoát vị não có thể gây thiểu năng trí tuệ, tuy nhiên, hầu hết người mắc hội chứng Knobloch không bị ảnh hưởng.
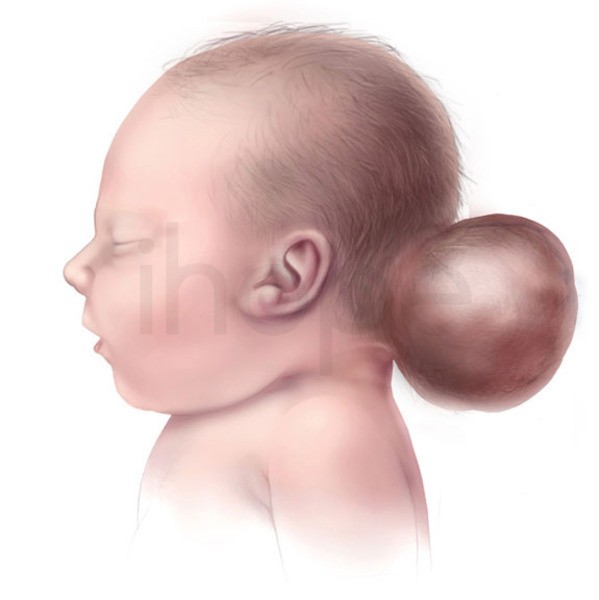
Ảnh: U não chẩm
Nguồn: U.S National Library of Medicine
Độ phổ biến
Hội chứng Knobloch hiếm gặp, do đó hiện nay tỷ lệ mắc bệnh vẫn chưa được thống kê cụ thể.
Nguyên nhân
Đột biến gen COL18A1 gây ra hội chứng Knobloch. Gen COL18A1 cung cấp hướng dẫn tạo ra protein collagen XVIII trong màng đáy của thuỷ tinh thể, võng mạc và các loại mô khác. Màng đáy có dạng tấm mỏng, nó ngăn cách và hỗ trợ tế bào trong các mô. Người ta chưa rõ chức năng của loại protein này, nhưng nó có liên quan đến quá trình phát triển bình thường của mắt.
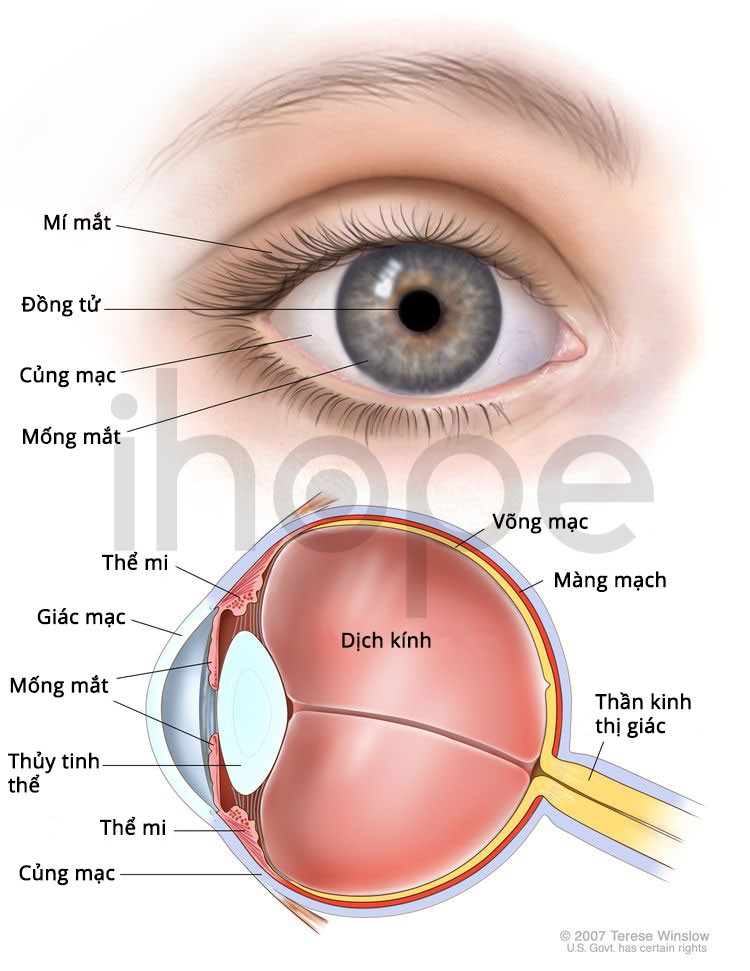
Nguồn: Terese Winslow
Đột biến gen COL18A1 dẫn đến phiên bản protein ngắn bất thường, từ đó gây ra các dấu hiệu và triệu chứng của hội chứng Knobloch. Tuy nhiên người ta chưa rõ cơ chế hình thành nên các triệu chứng.
Đột biến gen COL18A1 có thể gây ra hội chứng Knobloch loại I. Theo một số nghiên cứu, đột biến ít nhất hai gen khác chưa được xác định gây ra hội chứng Knobloch loại II và III. Tuy nguyên nhân di truyền khác nhau, ba loại đều có các dấu hiệu và triệu chứng tương tự.
Chẩn đoán
Knobloch syndrome thường được chẩn đoán dựa trên các dấu hiệu cụ thể của người bệnh. Một số xét nghiệm chuyên sâu có thể được chỉ định để chẩn đoán.
Các đặc điểm lâm sàng thường gặp bao gồm cận thị nặng, thoái hoá thủy tinh thể và võng mạc, các khuyết tật sọ. Bác sĩ có thể chỉ định chụp cộng hưởng từ (MRI), chụp CT xem người bệnh có phù não hay không, từ đó đánh giá kích thước và vị trí nếu có. Xét nghiệm di truyền tìm các đột biến gen COL18A1 liên quan đến hội chứng Knobloch thông qua mẫu máu hoặc mẫu phết niêm mạc miệng.
Điều trị
Hiện nay chưa có phương pháp điều trị hoàn toàn hội chứng Knobloch. Mục tiêu điều trị chủ yếu nhằm giảm thiểu triệu chứng và hỗ trợ chức năng của cơ quan bị ảnh hưởng. Người bệnh nên đeo kính để cải thiện tình trạng cận thị nặng. Trường hợp bong võng mạc hoặc khuyết tật đầu sọ có thể phẫu thuật chỉnh sửa. Bên cạnh đó, người bệnh cần thăm khám định kỳ nhằm đánh giá hiệu quả điều trị và phòng ngừa biến chứng có thể xảy ra.
Dạng di truyền
Hội chứng Knobloch di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh di truyền lặn trên nhiễm sắc thể thường sẽ có bố và mẹ mang một bản sao của gen đột biến, nhưng bố mẹ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Bệnh di truyền lặn đột biến gen COL18A1, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kỳ.
Các tên gọi khác
- Retinal detachment and occipital encephalocele
References
- Genetic Testing Information. Knobloch syndrome. Retrieved March 22, 2023 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C4551775/
- Genetic and Rare Diseases Information Center. Knobloch syndrome. Retrieved March 22, 2023 from https://rarediseases.info.nih.gov/diseases/380/knobloch-syndrome/
- Catalog of Genes and Diseases from OMIM. KNOBLOCH SYNDROME 1. Retrieved March 22, 2023 from https://omim.org/entry/267750
- U.S National Library of Medicine. Knobloch syndrome. Retrieved March 22, 2023 from https://medlineplus.gov/genetics/condition/knobloch-syndrome/
- American Academy of Ophthalmology. Knobloch syndrome. Retrieved March 22, 2023 from https://eyewiki.aao.org/Knobloch_Syndrome
- Frontiers. Case Report: Novel Biallelic Variants in the COL18A1 Gene in a Chinese Family With Knobloch Syndrome. Retrieved March 22, 2023 from https://www.frontiersin.org/articles/10.3389/fneur.2022.853918/full
- National Organization for Rare Disorders. Knobloch syndrome. Retrieved March 22, 2023 from https://rarediseases.org/gard-rare-disease/knobloch-syndrome/
- Orphanet. Knobloch syndrome. Retrieved March 22, 2023 from https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=1571
- Fukai N, Eklund L, Marneros AG, Oh SP, Keene DR, Tamarkin L, Niemela M, Ilves M, Li E, Pihlajaniemi T, Olsen BR. Lack of collagen XVIII/endostatin results in eye abnormalities. EMBO J. 2002 Apr 2;21(7):1535-44. doi: 10.1093/emboj/21.7.1535