
Hội chứng mất đoạn 2p15-p16.1 (2p15-p16.1 microdeletion syndrome) bắt nguồn từ mất đoạn ngắn trên nhiễm sắc thể số 2. Kích thước đoạn gen bị mất dao động từ 570 kilobase (kb) đến 5,7 megabase (Mb). Hội chứng này chủ yếu gây ra các bất thường liên quan đến phát triển thể chất và trí tuệ.
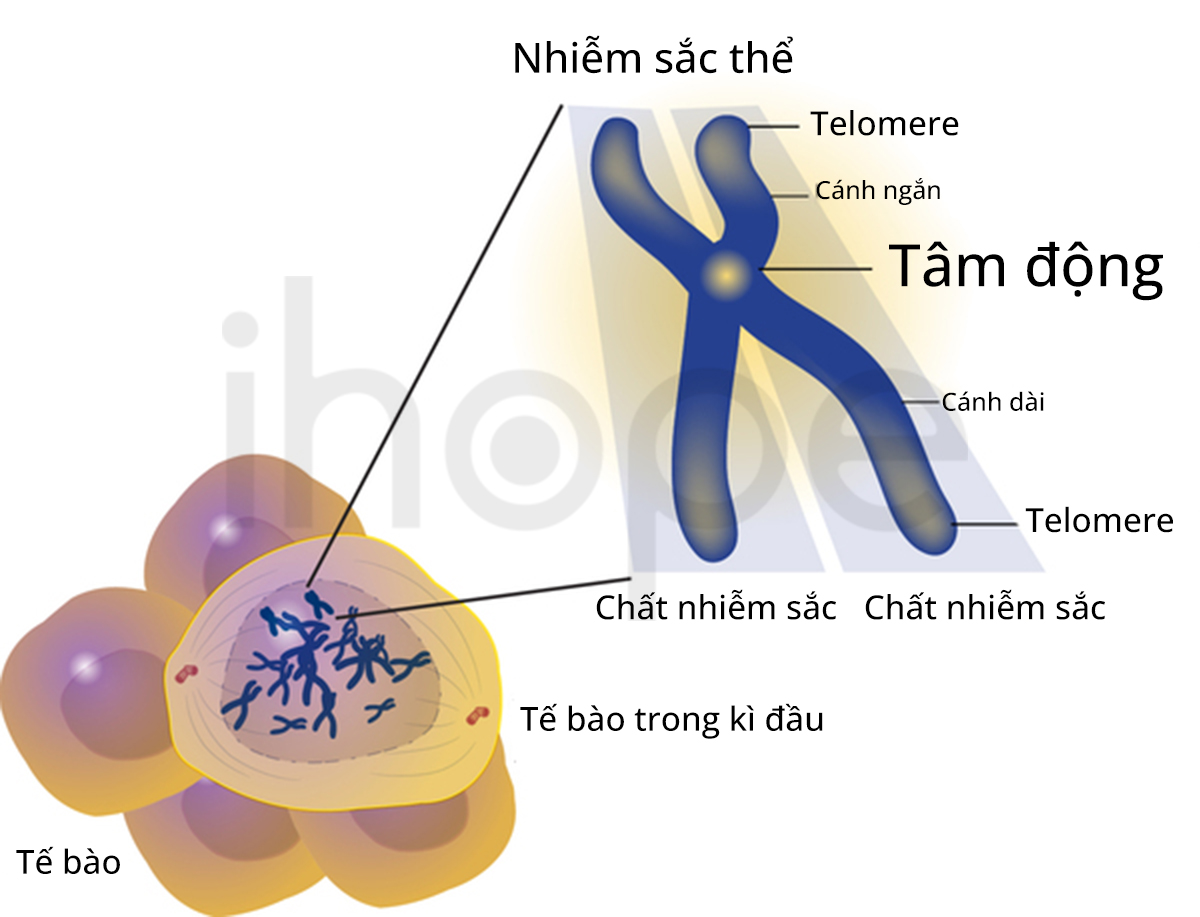
Nguồn: U.S. National Library of Medicine
Biểu hiện lâm sàng
Bệnh nhân mắc hội chứng mất đoạn 2p15-p16.1 thường có các điểm dị biệt trên khuôn mặt bao gồm:
- Đầu nhỏ
- Trán lõm
- Khoảng cách giữa hai mắt xa
- Nếp mí mắt rẻ quạt
- Sụp mi mắt
- Mũi lớn và gồ lên bất thường
- Nhân trung phẳng
- Miệng nhỏ, vòm miệng cao và hẹp
- Môi dưới lật ra ngoài
- Hàm dưới kém phát triển
Trẻ mắc bệnh thường thấp còi và thiểu năng trí tuệ mức độ nhẹ đến trung bình. Do đó, trẻ chậm phát triển ngôn ngữ, khả năng nói và gặp khó khăn trong học tập. Một số người bệnh có dấu hiệu của tự kỉ, động kinh hoặc rối loạn tăng động giảm chú ý.
Ngoài ra, bệnh nhân có thể bị teo hoặc thiểu sản dây thần kinh thị giác hai bên mắt, dẫn đến biểu hiện suy giảm thị lực.
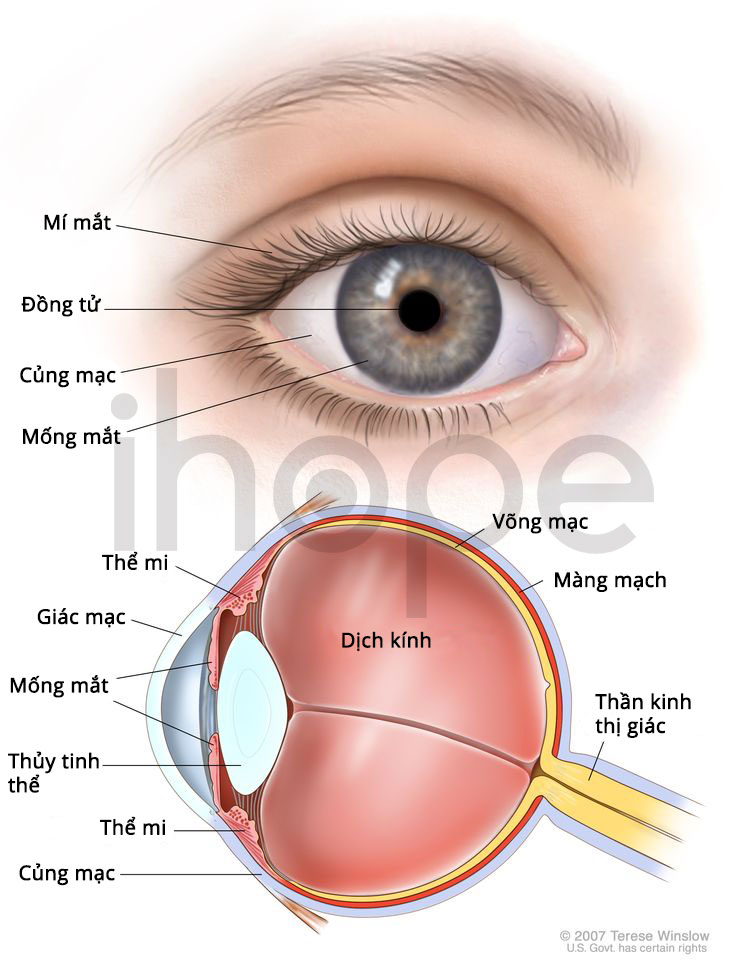
Nguồn: Medlineplus.gov
Một số người bệnh có bất thường liên quan đến hệ tiết niệu sinh dục và thận, ví dụ trẻ có thể bị nang ống mật chủ bẩm sinh. Mức độ nghiêm trọng cũng khác nhau giữa từng trường hợp.
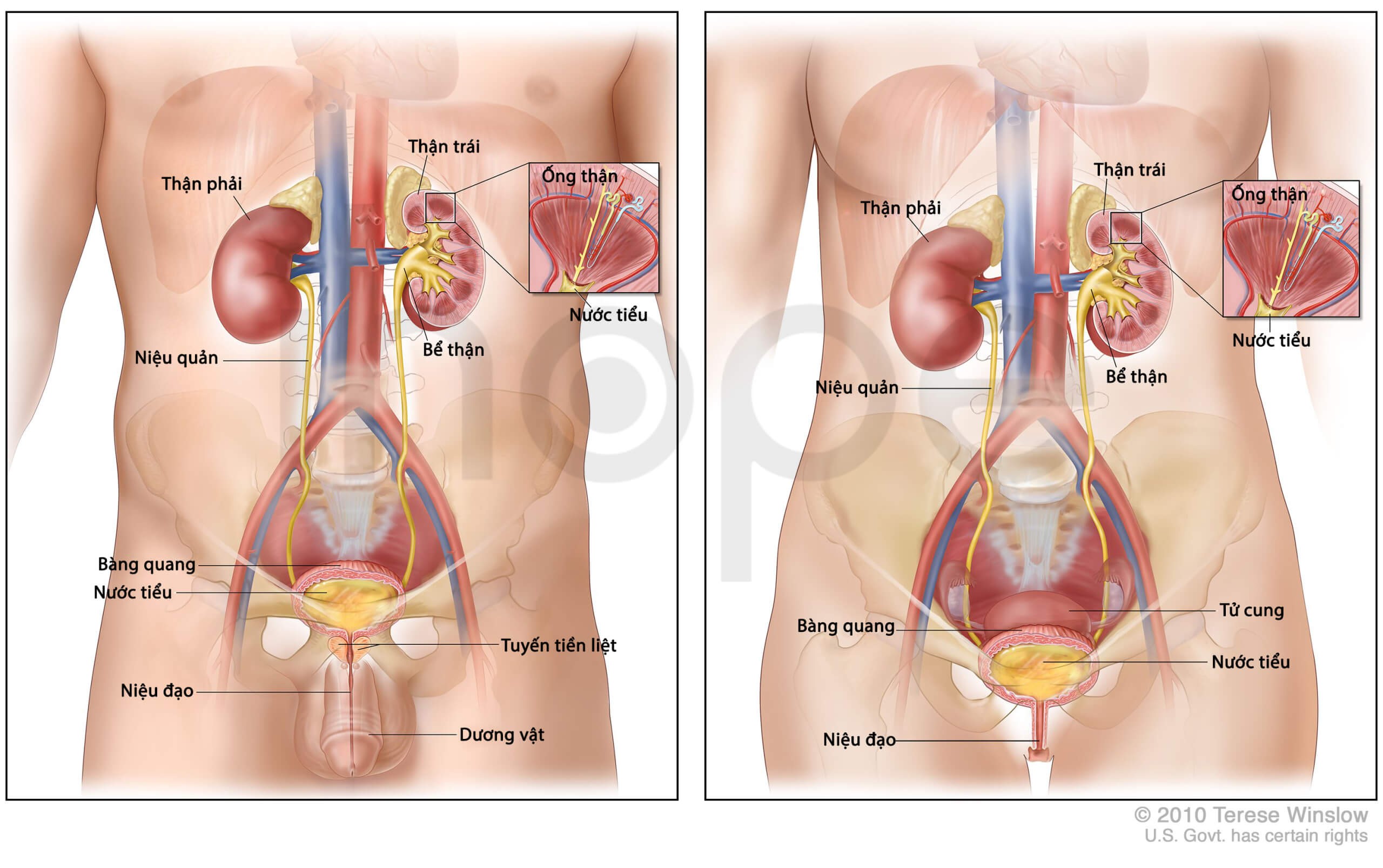
Nguồn: U.S. National Library of Medicine
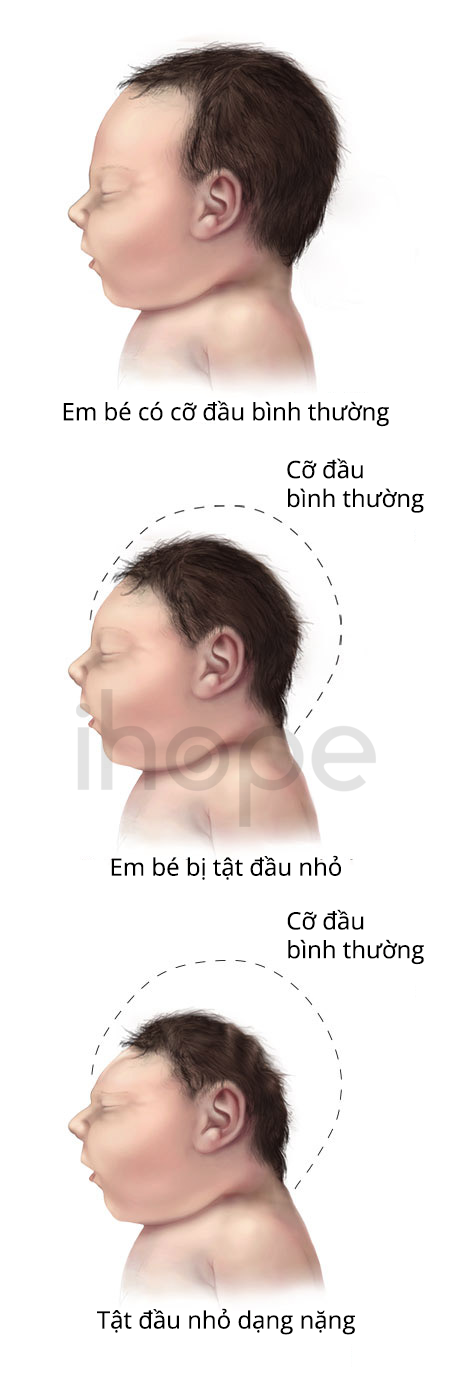
Ảnh: Tật đầu nhỏ
Nguồn: Centers for Disease Control and Prevention
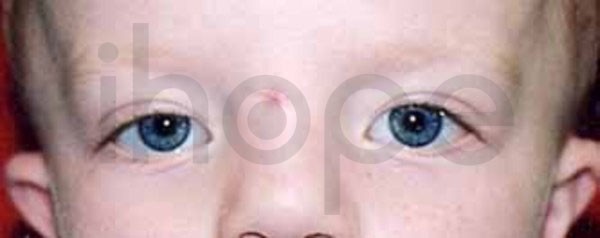
Ảnh: Khoảng cách giữa hai mắt rộng
Nguồn: Elements of Morphology, National Human Genome Research Institute
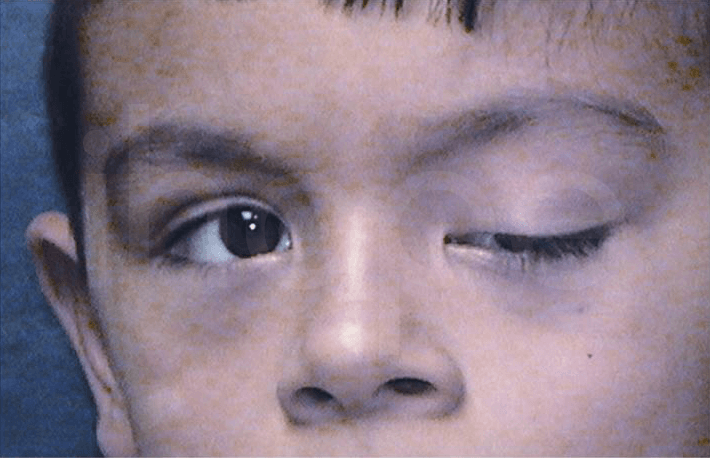
Ảnh: Mắt bị sụp mí
Nguồn: American Academy of Ophthalmology
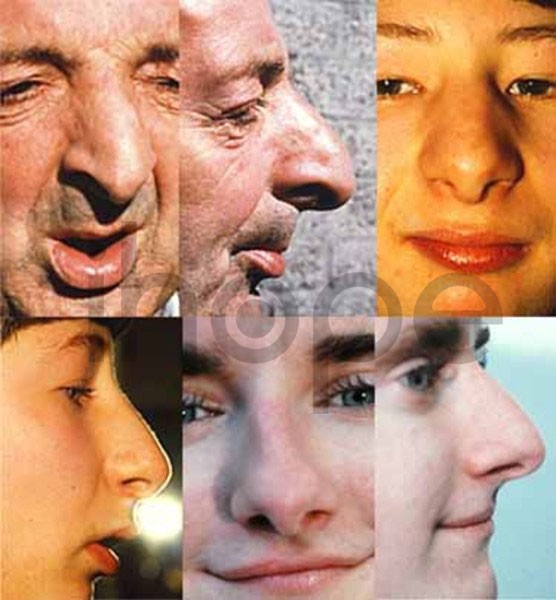
Ảnh: Cấu trúc mũi bất thường
Nguồn: Elements of Morphology, National Human Genome Research Institute
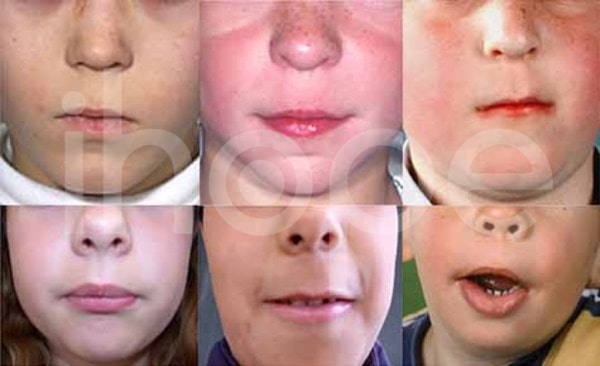
Ảnh: Không có nhân trung
Nguồn: Elements of Morphology, National Human Genome Research Institute
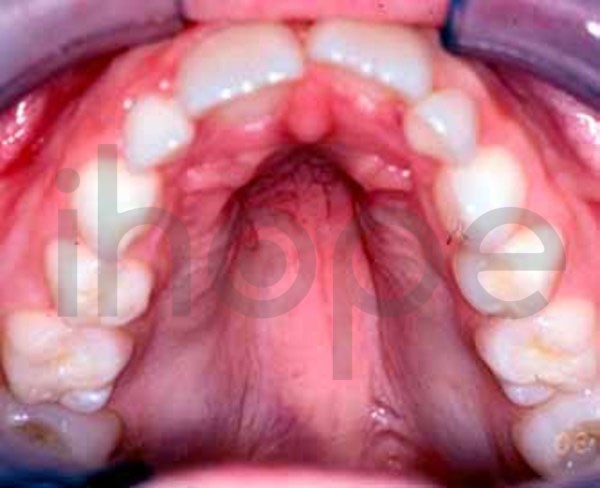
Ảnh: Vòm miệng cao
Nguồn: U.S. National Library of Medicine
Độ phổ biến
Hội chứng mất đoạn 2p15-p16.1 là bệnh di truyền hiếm gặp. Hiện nay, người ta mới ghi nhận khoảng 5 trường hợp mắc bệnh trên toàn thế giới.
Nguyên nhân
Mất đoạn giữa vùng 2p15 và 2p16.1 trên nhiễm sắc thể 2 là nguyên nhân chính gây ra hội chứng mất đoạn 2p15-p16.1. Đoạn bị mất có kích thước dao động từ 570 kb đến 5,7; nó mang nhiều gen quan trọng như XPO1, USP34, BCL11A, REL, PAPOLG, PEX13, COMMD1, B3GNT2, VRK2 và EHBP1. Trong đó, mất hai gen XPO1 và USP34 chiếm hơn 70% tổng số các trường hợp.
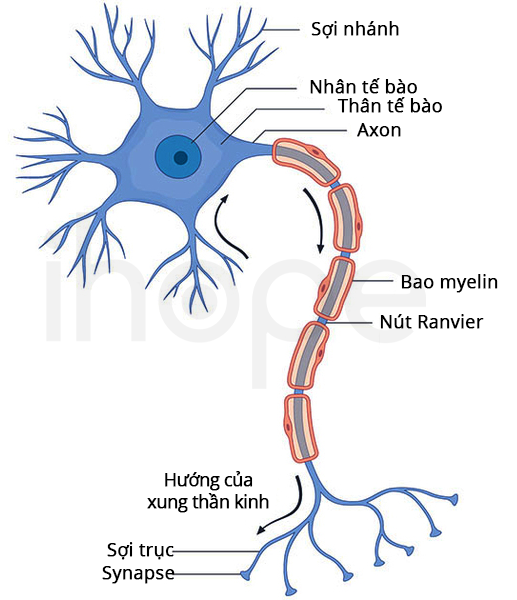
Gen USP34
Gen USP34 cung cấp hướng dẫn tạo ra enzyme ubiquitin-specific peptidase 34. Enzyme này tham gia điều chỉnh các hoạt động quan trọng trong cơ thể, bao gồm quá trình phát triển tế bào và duy trì độ ổn định mô.
Gen XPO1
Gen XPO1 cung cấp hướng dẫn tạo ra protein exportin 1. Protein này có chức năng vận chuyển các phân tử quan trọng ra khỏi nhân tế bào, hỗ trợ quá trình phát triển tế bào thần kinh và duy trì cấu trúc nhiễm sắc thể. Thiếu một bản sao của gen XPO1 cùng với những gen khác như BCL11A, REL và VRK2 góp phần gây ra triệu chứng bệnh. Tuy nhiên, hội chứng mất đoạn 2p15-p16.1 không phụ thuộc vào một gen duy nhất, bệnh chịu tác động từ tương tác giữa yếu tố di truyền và môi trường.
Nguồn: Eunice Kennedy Shriver National Institute of Child Health and Human Development
Chẩn đoán
Hội chứng mất đoạn 2p15-16.1 được chẩn đoán dựa trên các biểu hiện lâm sàng như khuôn mặt dị biệt, chậm phát triển và thiểu năng trí tuệ. Ngoài ra, người bệnh cần thực hiện xét nghiệm di truyền nhằm xác nhận kết quả chẩn đoán.
Hiện nay, phân tích vi mảng nhiễm sắc thể (Chromosomal Microarray Analysis - CMA) là phương pháp chính nhằm phát hiện đột biến mất vi đoạn, qua đó người ta có thể lập bản đồ chi tiết về những bất thường trên nhiễm sắc thể. Nếu kết quả CMA cho thấy có mất đoạn, kĩ thuật lai huỳnh quang tại chỗ (FISH) được sử dụng nhằm xác định vị trí cụ thể của đoạn mất trên nhiễm sắc thể 2.
Ngoài ra, bác sĩ có thể thu thập thông tin chi tiết về bệnh sử gia đình nhằm xác định mô hình di truyền hoặc thành viên trong gia đình có biểu hiện tương tự.
Điều trị
Hiện nay, chưa có phương pháp điều trị hoàn toàn hội chứng mất đoạn 2p15-p16.1. Các liệu pháp chủ yếu nhằm giảm nhẹ triệu chứng và cải thiện chất lượng đời sống người bệnh. Trẻ mắc bệnh cần điều trị phục hồi chức năng vận động, trị liệu ngôn ngữ và hành vi. Ngoài ra, bác sĩ có thể kê đơn thuốc để kiểm soát triệu chứng co giật của trẻ.
Dạng di truyền
Phần lớn các trường hợp mắc hội chứng mất đoạn 2p15-p16.1 không di truyền. Bệnh xảy ra do đột biến mới (de novo) xuất hiện trong quá trình hình thành tế bào sinh sản (trứng, tinh trùng) hoặc quá trình phát triển phôi sớm. Các trường hợp di truyền bệnh từ cha mẹ hiếm gặp hơn.
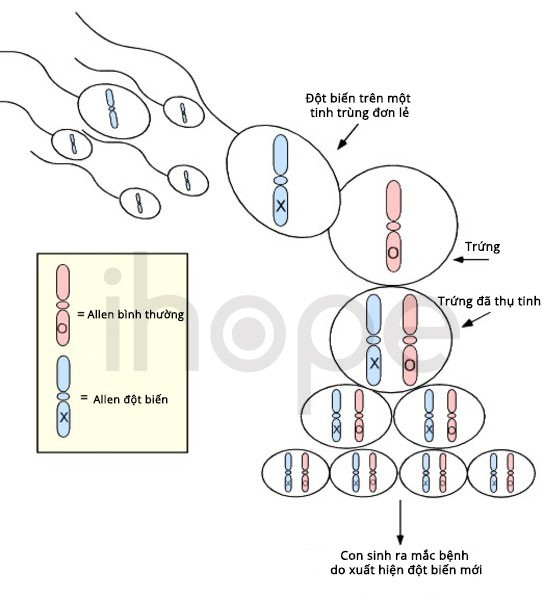
Nguồn: U.S. National Library of Medicine
Phòng ngừa
Hội chứng mất đoạn 2p15-p16.1 do đột biến ngẫu nhiên xảy ra trong quá trình tạo phôi, vì vậy tất cả thai phụ đều có nguy cơ mang thai bị bệnh. Do đó, sàng lọc phát hiện bệnh sớm vô cùng quan trọng và cần thiết. Sản phụ nên thăm khám và siêu âm định kì cũng như làm các xét nghiệm cần thiết như sàng lọc NIPT nhằm phát hiện sớm vấn đề có thể xảy ra với thai nhi.
Các tên gọi khác
- 2p15-p16.1 microdeletion syndrome
- 2p15p16.1 microdeletion syndrome
- Del(2)(p15p16.1)
- chromosome 2p16.1-p15 deletion syndrome
- chromosome 2p16.1-p15 deletion syndrome, isolated cases
- monosomy 2p15-p16.1
- monosomy 2p15p16.1
References
- Genetic Testing Information. Chromosome 2p16.1-p15 deletion syndrome. Retrieved November 18, 2024 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C2675875/
- Genetic and Rare Diseases Information Center. 2p15p16.1 microdeletion syndrome. Retrieved November 18, 2024 from https://rarediseases.info.nih.gov/diseases/13391/2p15p161-microdeletion-syndrome
- Catalog of Genes and Diseases from OMIM. CHROMOSOME 2p16.1-p15 DELETION SYNDROME. Retrieved November 18, 2024 from https://www.omim.org/entry/612513
- MalaCards. Chromosome 2p16.1-P15 Deletion Syndrome. Retrieved November 18, 2024 from https://www.malacards.org/card/chromosome_2p161_p15_deletion_syndrome
- National Institute of Health. Characteristics of 2p15-p16.1 microdeletion syndrome: Review and description of two additional patients. Retrieved November 18, 2024 from https://pubmed.ncbi.nlm.nih.gov/25900130/
- National Institute of Health. 2p15–p16.1 microdeletion syndrome: molecular characterization and association of the OTX1 and XPO1 genes with autism spectrum disorders. Retrieved November 18, 2024 from https://pmc.ncbi.nlm.nih.gov/articles/PMC3230356/
- National Organization for Rare Disorders. chromosome 2p16.1-p15 deletion syndrome. Retrieved November 18, 2024 from https://rarediseases.org/mondo-disease/chromosome-2p16-1-p15-deletion-syndrome/
- Orphanet. 2p15p16.1 microdeletion syndrome. Retrieved November 18, 2024 from https://www.orpha.net/en/disease/detail/261349





