Thiếu chất hoạt hóa GM2 (GM2 activator deficiency) là bệnh di truyền hiếm gặp. Bệnh thường gây ra tổn thương cho não và tiến triển nghiêm trọng theo thời gian.
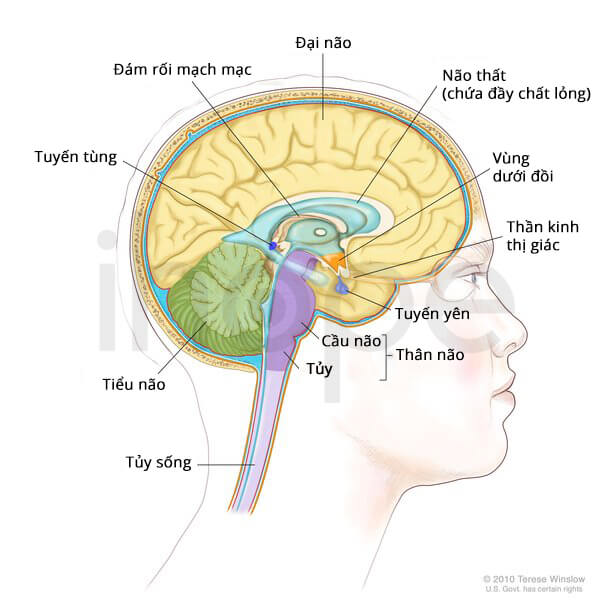
Nguồn: Terese Winslow
Biểu hiện lâm sàng
Đa số trẻ thiếu chất hoạt hóa GM2 mắc bệnh dạng cấp tính. Độ tuổi khởi phát bệnh vào khoảng 4–12 tháng tuổi. Trong khoảng thời gian này, trẻ phát triển chậm lại và các cơ vận động yếu dần. Tuổi thọ của bệnh nhân thường kéo dài đến hết giai đoạn trẻ nhỏ.

Nguồn: U.S National Library of Medicine
Những dấu hiệu phổ biến của bệnh gồm có:
- Chậm hoặc ngừng phát triển
- Yếu cơ
- Mất kĩ năng đã học trước đó như lật người, ngồi và bò
- Phản ứng quá mức với tiếng động lớn
- Động kinh
- Mất thị lực
- Thiểu năng trí tuệ
- Không phản ứng với môi trường xung quanh
Ngoài ra, trẻ bệnh còn xuất hiện đốm đỏ anh đào trong mắt—triệu chứng phổ biến của bệnh thiếu chất hoạt hóa GM2 dạng trẻ sơ sinh cấp tính.
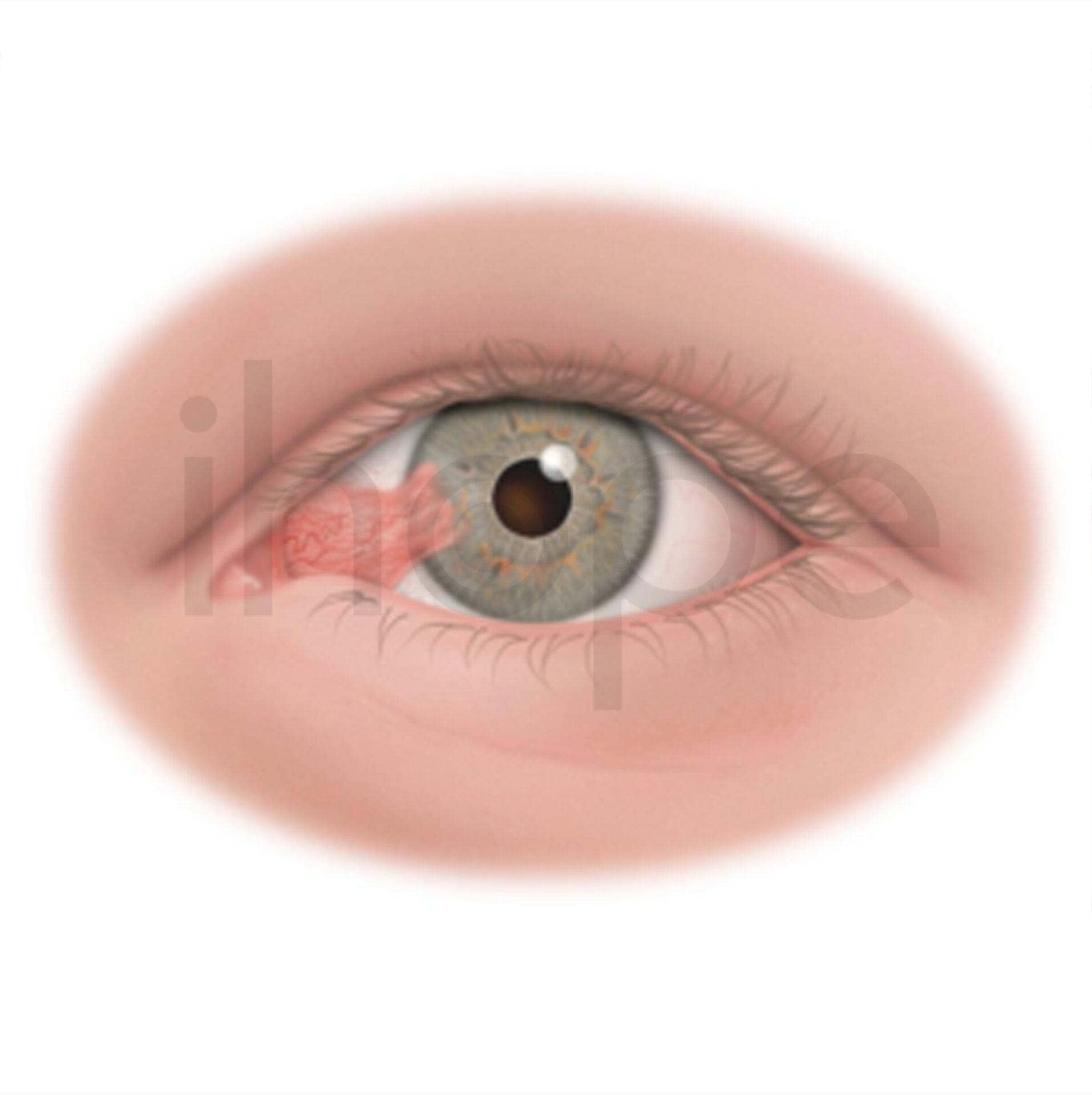
Nguồn: VisionCenter
Trong một số trường hợp, bệnh nhân có triệu chứng nhẹ và thời gian khởi phát muộn. Bệnh dạng khởi phát muộn chưa xác định độ phổ biến vì nó rất hiếp gặp.
Độ phổ biến
Bệnh thiếu chất hoạt hóa GM2 rất hiếm gặp, chưa đến 30 bệnh nhân được ghi nhận trên toàn thế giới.
Nguyên nhân
Đột biến gen GM2A gây ra bệnh thiếu chất hoạt hóa GM2. Gen GM2A cung cấp hướng dẫn tạo ra protein hoạt hóa GM2 ganglioside có chức năng kích hoạt enzyme beta-hexosaminidase A. Enzyme beta-hexosaminidase A và protein hoạt hóa GM2 ganglioside hoạt động cùng nhau trong lysosome—bào quan phân giải và tái chế nhiều loại phân tử khác nhau. Trong lysosome, protein hoạt hóa liên kết với chất béo GM2 ganglioside rồi đưa nó cho enzyme beta-hexosaminidase A phân giải.
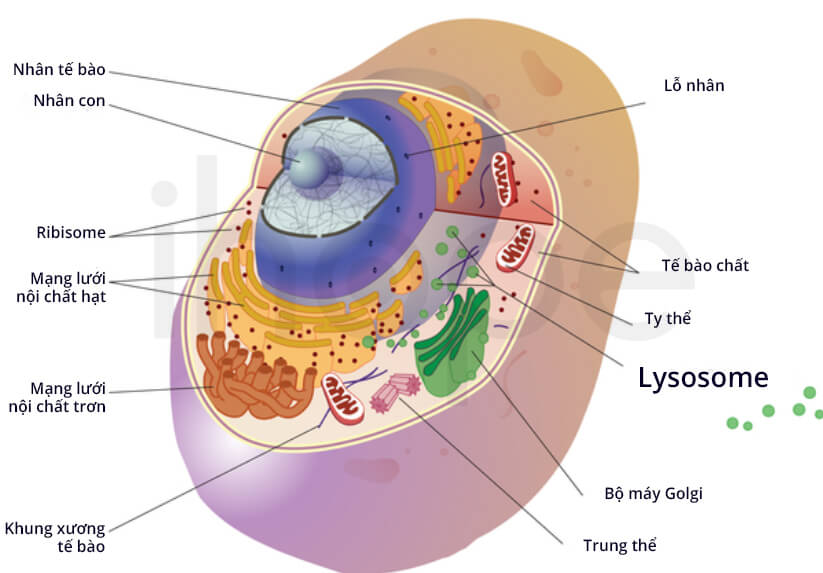
Nguồn: Darryl Leja, NHGRI
Đột biến gen GM2A ức chế hoạt động của protein hoạt hóa GM2 ganglioside nên quá trình phân giải chất béo GM2 ganglioside của enzyme beta-hexosaminidase A bị gián đoạn. Do đó, lượng GM2 ganglioside trong cơ thể tăng lên và tích tụ chủ yếu trong hệ thần kinh, cuối cùng những triệu chứng của bệnh thiếu chất hoạt hóa GM2 khởi phát.
Tình trạng thiếu chất hoạt hóa GM2 khiến enzyme giảm hoạt động trong lysosome và GM2 ganglioside tích tụ. Vì vậy, bệnh này còn gọi là bệnh tích trữ lysosome hay bệnh GM2 gangliosidosis.
Chẩn đoán
Bệnh thiếu chất hoạt hóa GM2 được chẩn đoán dựa trên triệu chứng lâm sàng như mắt có đốm đỏ, yếu cơ, động kinh, thiếu năng trí tuệ. Ngoài ra, bác sĩ cũng chẩn đoán dựa trên bệnh sử gia đình của bệnh nhân. Mặt khác, bệnh nhân nên chụp MRI não nhằm kiểm tra lượng myelin và tín hiệu T2 trong chất trắng dưới vỏ não, hạch nền hoặc đồi thị. Khi cần thiết, người bệnh có thể thực hiện xét nghiệm di truyền để xác nhận chẩn đoán.
Các phương pháp xét nghiệm di truyền phổ biến bao gồm:
- Xét nghiệm gen nhắm mục tiêu: xét nghiệm đơn gen, bảng đa gen
- Xét nghiệm toàn bộ bộ gen: giải trình tự exome, giải trình tự bộ gen
Điều trị
Hiện nay, chưa có liệu pháp điều trị hoàn toàn bệnh thiếu chất hoạt hóa GM2. Các liệu pháp tập trung làm giảm triệu chứng và cải thiện cuộc sống cho người bệnh.
Các phương pháp bao gồm:
- Cung cấp đủ dinh dưỡng và nước
- Kiểm soát bệnh truyền nhiễm
- Bảo vệ đường hô hấp
- Giảm tần suất động kinh
Ngoài ra, người bệnh cần thực hiện kiểm tra sức khỏe định kì nhằm điều chỉnh hoặc bổ sung thêm phương pháp hỗ trợ phù hợp.
Dạng di truyền
Bệnh thiếu chất hoạt hóa GM2 di truyền theo kiểu lặn trên nhiễm sắc thể thường, nghĩa là cả hai bản sao của gen trong mỗi tế bào đều có đột biến. Cha mẹ của một cá nhân mắc bệnh lặn trên nhiễm sắc thể thường đều mang một bản sao của gen đột biến, nhưng họ thường không biểu hiện dấu hiệu và triệu chứng của bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Bệnh thiếu chất hoạt hóa GM2 di truyền lặn đột biến gen GM2A, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kì.
Các tên gọi khác
- GM2 gangliosidosis, AB variant
- Hexosaminidase activator deficiency
- Tay-Sachs disease, AB variant
References
- Genetic Testing Information. Tay-Sachs disease, variant AB. Retrieved September 09, 2024 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0268275/
- Genetic and Rare Diseases Information Center. GM2-gangliosidosis, B, B1, AB variant. Retrieved September 09, 2024 from https://rarediseases.info.nih.gov/diseases/2522/index
- Catalog of Genes and Diseases from OMIM. GM2-GANGLIOSIDOSIS, AB VARIANT. Retrieved September 09, 2024 from https://omim.org/entry/272750
- U.S. National Library of Medicine. GM2 activator deficiency. Retrieved September 09, 2024 from https://medlineplus.gov/genetics/condition/gm2-activator-deficiency/
- National Institute of Health. GM2 Activator Deficiency. Retrieved September 09, 2024 from https://www.ncbi.nlm.nih.gov/books/NBK583219/
- National Institute of Health. GM2 Activator Deficiency Caused by a Homozygous Exon 2 Deletion in GM2A. Retrieved September 09, 2024 from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5874204/
- Europe PMC. GM2 Activator Deficiency. Retrieved September 09, 2024 from https://europepmc.org/article/nbk/nbk583219
- BioMed Central. GM2 gangliosidosis AB variant: novel mutation from India – a case report with a review. Retrieved September 09, 2024 from https://bmcpediatr.biomedcentral.com/articles/10.1186/s12887-016-0626-6





