Hội chứng Saul-Wilson (Saul-Wilson syndrome) là bệnh di truyền hiếm gặp tác động đến quá trình phát triển của xương với một loạt các triệu chứng đặc trưng. Đa số người bệnh có tầm vóc thấp bé. Cụ thể, trẻ bệnh thường chậm phát triển khi còn trong bụng mẹ. Khi trưởng thành, chiều cao trung bình của trẻ chỉ khoảng 107cm. Các biểu hiện của bệnh khởi phát từ trước khi sinh và tiếp tục kéo dài đến sau khi chào đời.
Biểu hiện lâm sàng
Ngoài vóc dáng thấp bé, hội chứng Saul-Wilson còn gây ra những đặc điểm dị biệt trên khuôn mặt bao gồm:
- Trán nhô ra phía trước
- Tóc và lông mày thưa
- Tĩnh mạch da đầu nổi rõ
- Sống mũi hẹp
- Mũi khoằm
- Trụ mũi rộng
- Môi trên mỏng
- Hàm dưới nhỏ
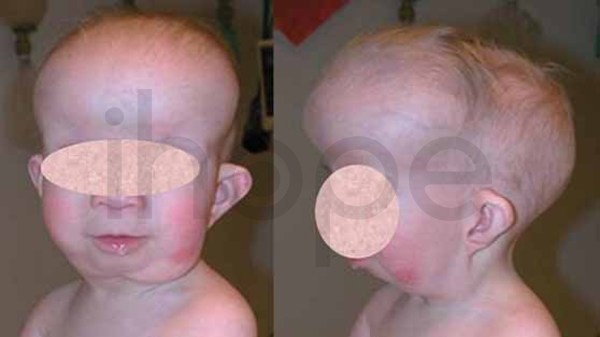
Ảnh: Trán nhô ra phía trước
Nguồn: Elements of Morphology, National Human Genome Research Institute
Ảnh: Tóc bệnh nhân mỏng và thưa hơn so với bình thường
Nguồn: MedlinePlus
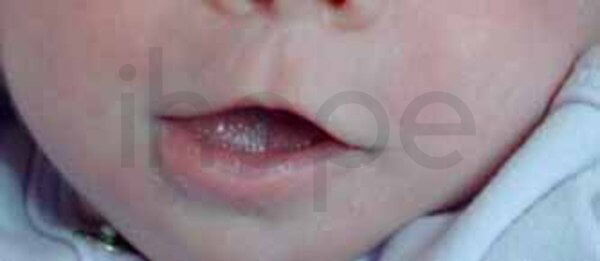
Ảnh: Môi trên mỏng
Nguồn: National Human Genome Research Institute
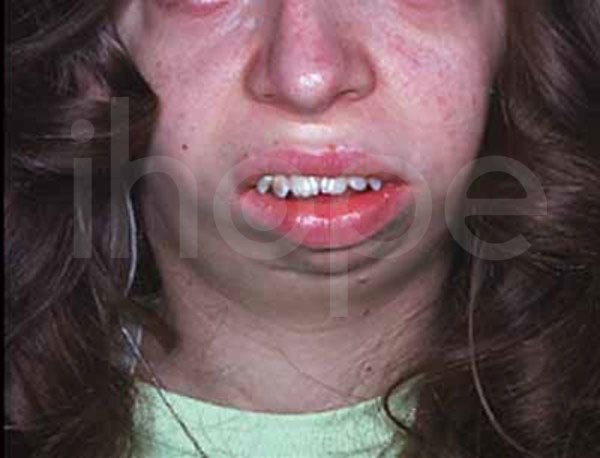
Ảnh: Xương hàm và xương cằm nhỏ, thu hẹp
Nguồn: U.S. National Library of Medicine
Những đặc điểm dị biệt này chủ yếu xuất hiện trên trẻ sơ sinh và khiến người bệnh có vẻ ngoài già trước tuổi.
Bên cạnh những bất thường về khuôn mặt, bệnh nhân cũng gặp phải các vấn đề về xương bao gồm:
- Xương dài phát triển bất thường
- Ngón tay và ngón chân ngắn
- Bàn chân khoèo
- Khớp háng dị dạng
- Xương cột sống dẹt
- Một số dị tật cột sống khác

Ảnh: Bàn chân khoèo
Nguồn: U.S. National Library of Medicine
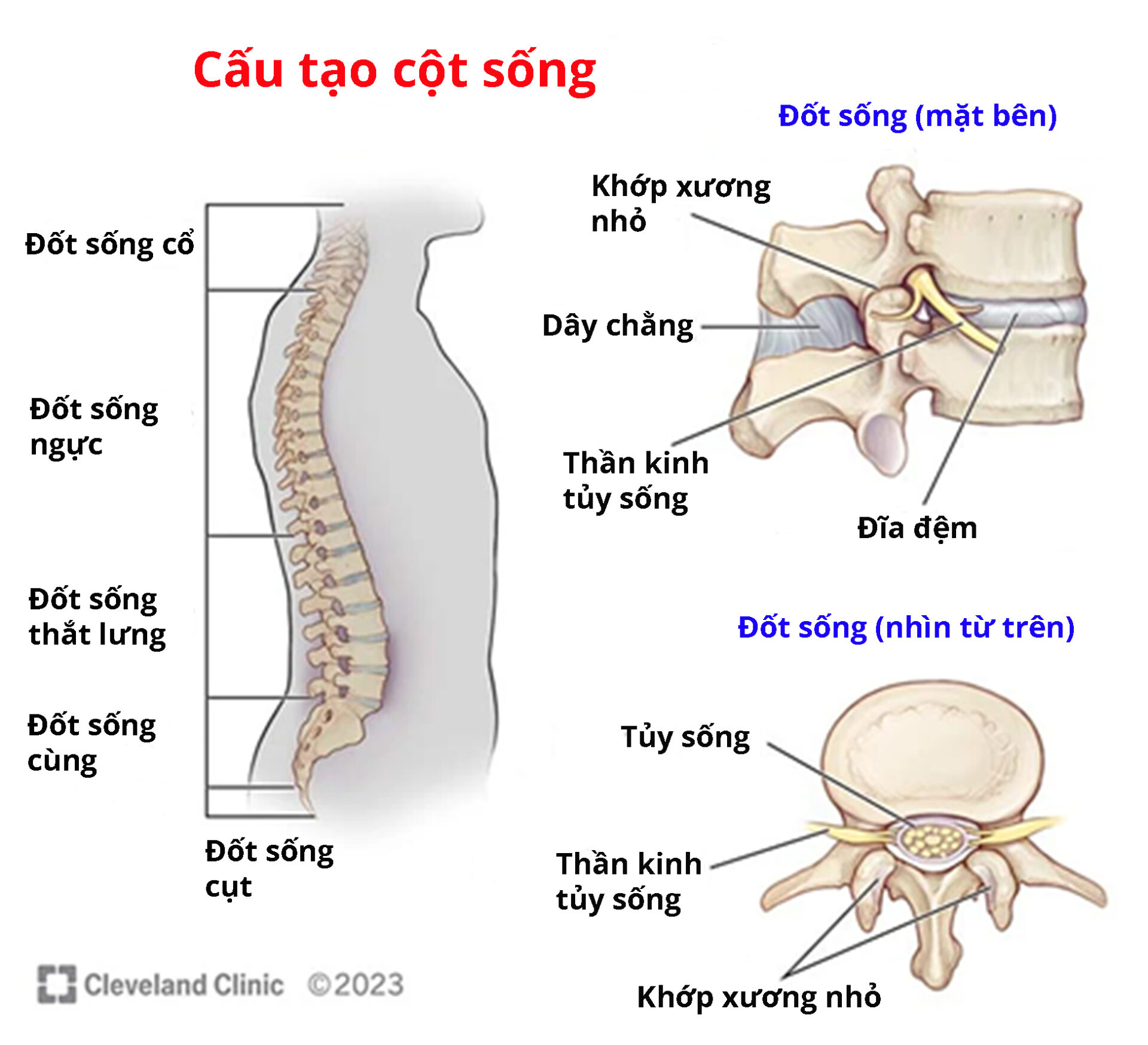
Nguồn: Cleverlandclinic.org
Ngoài ra, xương người bệnh cũng thường giòn hơn bình thường và dễ gãy dù chỉ va chạm nhẹ. Khi trưởng thành, người bệnh có thể xuất hiện triệu chứng đau khớp do thoái hóa khớp.
Một số vấn đề sức khỏe khác liên quan đến hội chứng Saul-Wilson bao gồm mất thính lực, đục thủy tinh thể, lòng trắng mắt có màu xanh, viêm võng mạc sắc tố có khả năng gây mất thị lực, chậm nói và chậm phát triển vận động. Tuy nhiên, trí thông minh của bệnh nhân thường không bị ảnh hưởng. Một số trẻ mắc bệnh giảm bạch cầu trung tính tái phát làm tăng nguy cơ nhiễm trùng đường hô hấp.
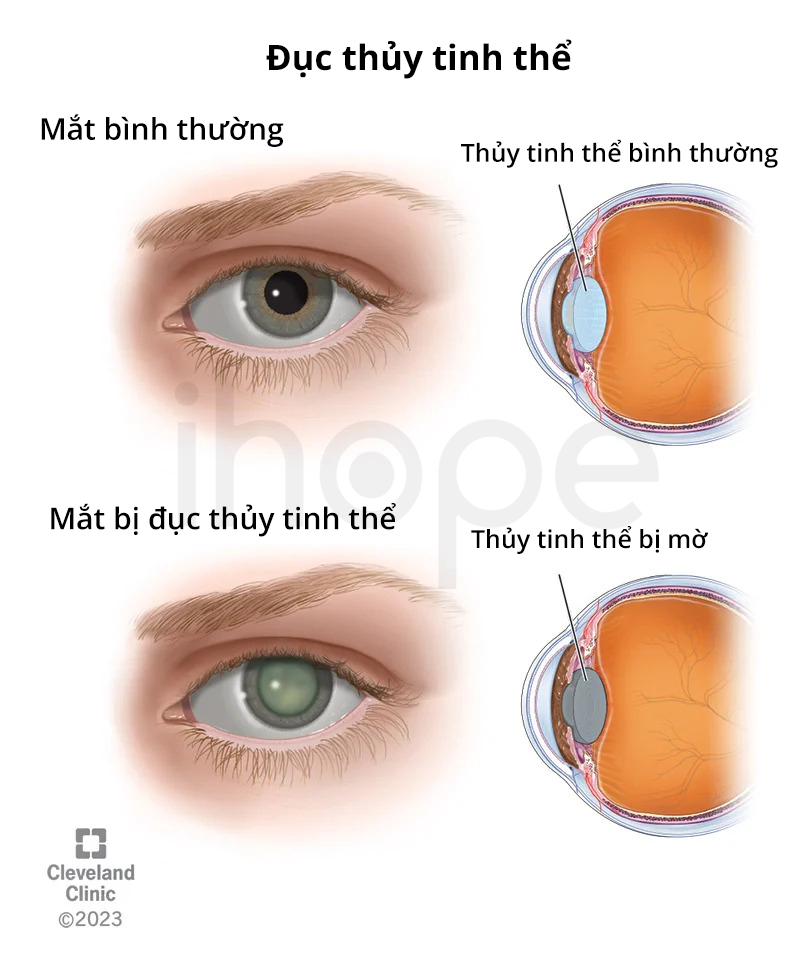
Nguồn: Cleveland Clinic
Độ phổ biến
Hội chứng Saul-Wilson rất hiếm gặp. Đến nay, 16 trường hợp đã được ghi nhận.
Nguyên nhân
Đột biến gen COG4 gây ra hội chứng Saul-Wilson. Gen này hướng dẫn sản xuất protein có vai trò quan trọng trong phức hợp COG. Phức hợp này hoạt động trong bộ máy Golgi—cơ quan của tế bào có chức năng xử lí và vận chuyển protein đến các vị trí khác nhau bao gồm cả lưới nội chất . Quá trình vận chuyển protein từ bộ máy Golgi đến lưới nội chất rất quan trọng để tái chế và sắp xếp lại các protein.
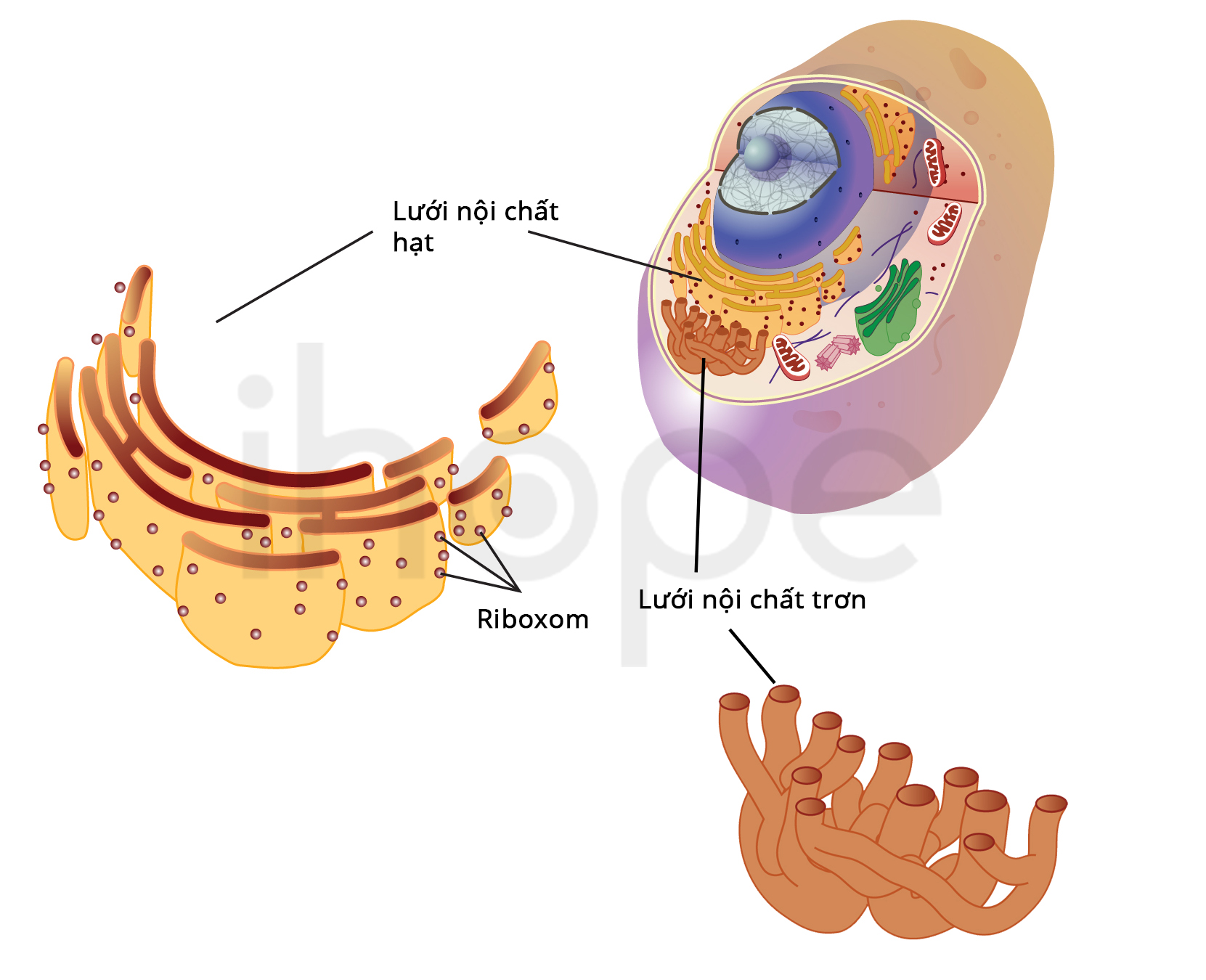
Ảnh: Lưới nội chất
Lưới nội chất là cấu trúc bên trong tế bào tham gia vào quá trình xử lý và vận chuyển protein
Nguồn: National Human Genome Research Institute
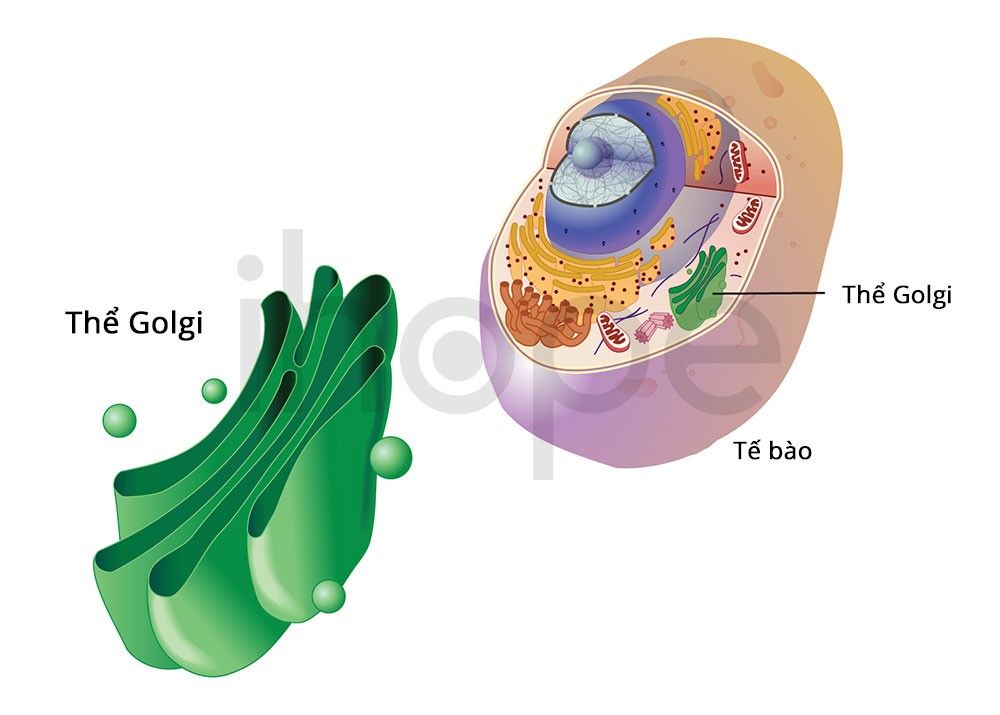
Ảnh: National Human Genome Research Institute
Đột biến gen COG4 khiến protein COG4 hoạt động bất thường nên quá trình vận chuyển protein giữa bộ máy Golgi và lưới nội chất tăng lên. Protein COG4 vẫn tham gia vào phức hợp COG nhưng các thay đổi này ảnh hưởng đến quá trình phát triển của xương và dẫn đến các triệu chứng của hội chứng Saul-Wilson. Cơ chế chính xác của quá trình này chưa được xác định.
Chẩn đoán
Hội chứng Saul-Wilson được chẩn đoán dựa trên biểu hiện lâm sàng kết hợp với xét nghiệm di truyền. Bác sĩ sẽ xác định các dấu hiệu đặc trưng như tầm vóc thấp bé, dị tật xương và các đặc điểm dị biệt trên khuôn mặt. Các xét nghiệm di truyền được sử dụng nhằm phát hiện đột biến gen COG4.
Điều trị
Hiện tại, chưa có phương pháp điều trị đặc hiệu cho hội chứng Saul-Wilson. Các liệu pháp tập trung vào quản lí triệu chứng và cải thiện chất lượng cuộc sống cho người bệnh.
Bệnh nhân có biểu hiện liên quan đến xương như bàn chân khoèo, trật khớp đốt sống cổ C1-C2, đau khớp và vận động khó khăn cần được theo dõi và điều trị bởi bác sĩ có chuyên môn về chỉnh hình. Mặt khác, các phương pháp bao gồm vật lí trị liệu và phục hồi chức năng có vai trò quan trọng đối với quá trình cải thiện khả năng vận động và giảm đau. Trong một số trường hợp, bác sĩ sẽ chỉ định bệnh nhân thực hiện phẫu thuật nhằm giải quyết các vấn đề nghiêm trọng như chèn ép tủy sống.
Đối với bệnh nhân đục thủy tinh thể và thoái hóa võng mạc, bác sĩ sẽ chỉ định điều trị bằng các phương pháp như thuốc nhỏ mắt, phẫu thuật hoặc sử dụng kính nhìn đêm và kính lọc bước sóng ánh sáng nhằm hỗ trợ thị lực. Bệnh nhân mất thính lực có thể sử dụng máy trợ thính hoặc ống thông khí tùy thuộc vào mức độ nghiêm trọng và nguyên nhân của triệu chứng.
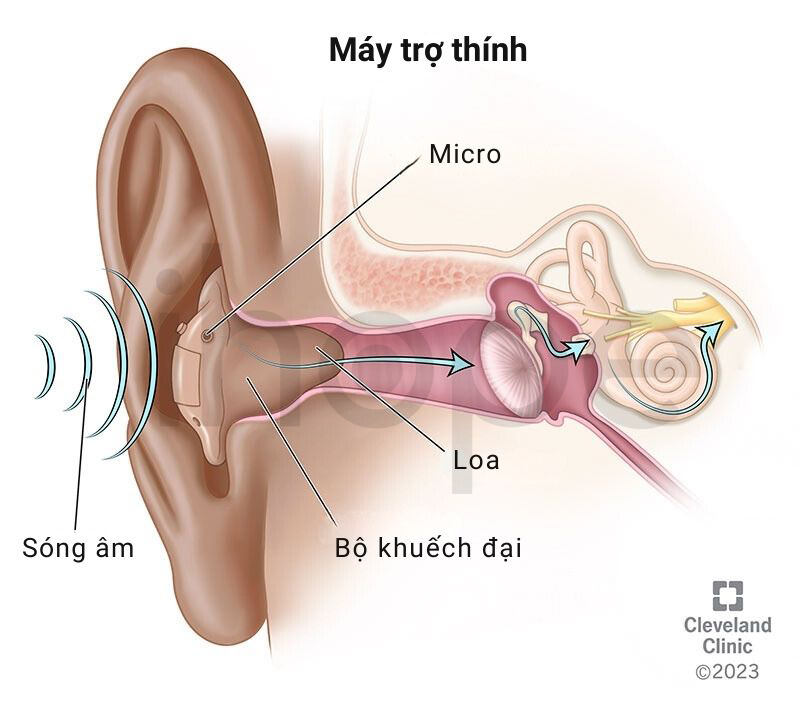
Nguồn: Cleveland Clinic
Ngoài ra, theo dõi thường xuyên quá trình tăng trưởng, phát triển vận động và ngôn ngữ của trẻ cũng rất quan trọng. Đối với trẻ có triệu chứng nhiễm trùng đường hô hấp tái phát do giảm bạch cầu trung tính, bác sĩ điều trị bằng kháng sinh và các biện pháp hỗ trợ khác. Trong một số trường hợp, bác sĩ có thể xem xét sử dụng yếu tố kích thích bạch cầu hạt (G-CSF) nhằm tăng số lượng bạch cầu trung tính và cải thiện khả năng chống nhiễm trùng của cơ thể.
Dạng di truyền
Hội chứng Saul-Wilson di truyền theo kiểu trội trên nhiễm sắc thể thường, nghĩa là chỉ cần một bản sao gen bị đột biến là đủ để gây bệnh. Phần lớn các trường hợp là do đột biến mới (de novo) xảy ra ngẫu nhiên trong quá trình hình thành tế bào sinh sản của bố mẹ hoặc trong giai đoạn phát triển phôi thai sớm.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Hội chứng Saul-Wilson di truyền trội trên nhiễm sắc thể thường, nếu cha hoặc mẹ mắc bệnh, con sinh ra sẽ có 50% khả năng di truyền bệnh. Do đó, để bảo đảm 100% khả năng con không bị bệnh, cha mẹ có thể chọn phương pháp thụ tinh nhân tạo IVF và sàng lọc phôi PGS/PGD. Các thành viên trong gia đình nên khám sức khỏe và tầm soát bệnh định kỳ nếu có thành viên mắc bệnh. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh.
Các tên gọi khác
- Microcephalic osteodysplastic dysplasia
- Microcephalic osteodysplastic dysplasia Saul Wilson type
References
- Genetic Testing Information. Microcephalic osteodysplastic dysplasia, Saul-Wilson type. Retrieved December 18, 2024 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C1300285/
- Catalog of Genes and Diseases from OMIM. SAUL-WILSON SYNDROME; SWILS. Retrieved December 18, 2024 from https://omim.org/entry/618150
- U.S. National Library of Medicine. Saul-Wilson syndrome. Retrieved December 18, 2024 from https://medlineplus.gov/genetics/condition/saul-wilson-syndrome/
- Frontiers. A Dominant Heterozygous Mutation in COG4 Causes Saul–Wilson Syndrome, a Primordial Dwarfism, and Disrupts Zebrafish Development via Wnt Signaling. Retrieved December 18, 2024 from https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2021.720688/full
- MalaCards. Saul-Wilson Syndrome (SWILS). Retrieved December 18, 2024 from https://www.malacards.org/card/saul_wilson_syndrome
- National Institute of Health. Growth in individuals with Saul–Wilson syndrome. Retrieved December 18, 2024 from https://pmc.ncbi.nlm.nih.gov/articles/PMC9016779/
- Orphanet. Microcephalic osteodysplastic dysplasia, Saul-Wilson type. Retrieved December 18, 2024 from https://www.orpha.net/en/disease/detail/85172