
Hội chứng Trichorhinophalangeal loại II (Trichorhinophalangeal syndrome type II) là bệnh gây dị dạng xương và khớp. Tên của bệnh mô tả một số vùng trên cơ thể bị ảnh hưởng, bao gồm tóc (tricho-), mũi (rhino-), ngón tay và ngón chân (phalangeal). Người bệnh có các biểu hiện đặc trưng bao gồm khuôn mặt dị biệt, thiểu năng trí tuệ và các bất thường khác.
Biểu hiện lâm sàng
Người bệnh thường xuất hiện nhiều khối u lành tính trong xương (u xương sụn). Chúng hình thành từ giai đoạn sơ sinh và ngừng phát triển vào khoảng tuổi thanh niên. Tùy thuộc vào vị trí, số lượng khối u xương mà bệnh nhân có các triệu chứng:
- Đau nhức
- Giới hạn phạm vi cử động khớp
- Tổn thương mạch máu, tủy sống
- Giảm mật độ khoáng trong xương gây loãng xương
- Chậm phát triển, tầm vóc thấp
- Đầu xương ngón tay, ngón chân hình nón
- Móng tay , móng chân mỏng, mọc ngược
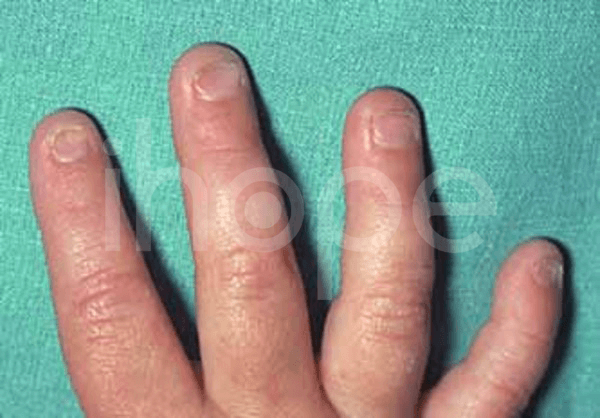
Ảnh: Móng tay mỏng
Nguồn: Elements of Morphology, National Human Genome Research Institute
Trẻ mắc bệnh có phạm vi cử động khớp lớn bất thường (chứng người dẻo). Nhưng khi u xương phát triển, khớp bắt đầu cứng lại và giảm khả năng vận động. Lệch khớp háng (loạn sản xương hông) thường thấy ở người bệnh trưởng thành.

Ảnh: Khớp ngón tay linh động
Nguồn: Mitskevich Uladzimir/Shutterstock.com
Trẻ mắc hội chứng Trichorhinophalangeal loại II bị thiểu năng trí tuệ nặng và có vẻ ngoài đặc trưng bao gồm:
- Tật đầu nhỏ, cằm nhỏ và tụt lại sau
- Hai mắt cách xa nhau hoặc mắt lác ẩn ngoài
- Lông mày rậm, mũi rộng với đầu mũi tròn, nhân trung rộng , môi trên mỏng
- Răng nhỏ, thiếu hoặc thừa răng
- Tóc thưa, mỏng, dễ gãy hoặc hói ở nam giới sau tuổi dậy thì
- Chứng tăng tiết mồ hôi
- Suy giảm thính lực
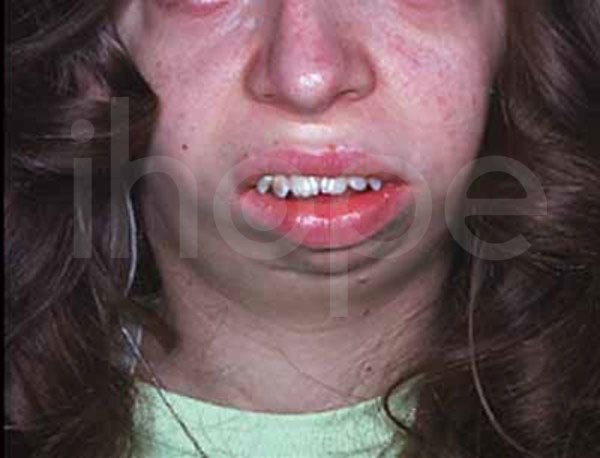
Ảnh: Xương hàm và xương cằm nhỏ, thu hẹp
Nguồn: U.S. National Library of Medicine
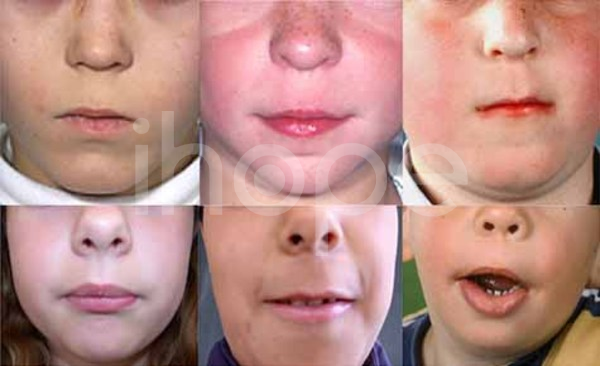
Ảnh: Vùng nhân trung rộng
Nguồn: U.S National Library of Medicine
Độ phổ biến
Hội chứng Trichorhinophalangeal loại II là bệnh hiếm gặp. Tỷ lệ mắc bệnh ở nam giới cao gấp ba lần so với nữ giới. Hơn 60 trường hợp đã được ghi nhận trên toàn thế giới.
Nguyên nhân
Hội chứng Trichorhinophalangeal loại II xảy ra do mất nhánh dài (q) của nhiễm sắc thể số 8 dẫn đến xóa các gen TRPS1, EXT1 và RAD21. Những gen này nắm vai trò điều hòa hoạt động gen, chức năng protein và phân chia tế bào.
- Gen TRPS1 cung cấp hướng dẫn tạo ra protein có chức năng điều hòa phiên mã. Mất gen TRPS1 làm giảm số lượng protein TRPS1, từ đó làm suy yếu chức năng điều hòa của một số gen kiểm soát quá trình phát triển của xương và sụn. Do đó, người bệnh có tầm vóc thấp, xương ngón tay và ngón chân bất thường, khớp dị dạng và khuôn mặt đặc trưng.
- Gen EXT1 cung cấp hướng dẫn tạo ra protein exostosin-1 trong bộ máy Golgi của tế bào. Protein exostosin-1 điều chỉnh các enzyme mới được sản xuất và các protein khác. Mất gen EXT1 gây thiếu hụt protein exostosin-1, cuối cùng dẫn đến hình thành u xương.
- Gen RAD21 cung cấp hướng dẫn tạo ra một loại protein tham gia điều hòa cấu trúc và tổ chức của nhiễm sắc thể trong quá trình phân bào. Mất gen RAD21 khiến các tế bào sản xuất lượng protein RAD21 ít hơn bình thường, từ đó có thể góp phần gây ra thiểu năng trí tuệ nhưng cơ chế vẫn chưa rõ ràng.
Kích thước của đoạn bị mất khác nhau giữa các bệnh nhân. Đoạn bị mất càng lớn, thông tin di truyền bị mất càng nhiều. Ngoài ba gen trên, các gen lân cận bị mất cũng góp phần gây ra thêm các triệu chứng khác của bệnh.
Trường hợp mất đoạn trên nhiễm sắc thể số 8 chỉ ảnh hưởng đến gen TRPS1, gọi là hội chứng Trichorhinophalangeal loại I. Người bệnh có triệu chứng tương tự như hội chứng Trichorhinophalangeal loại II nhưng không xuất hiện u xương và thiểu năng trí tuệ.
Chẩn đoán
Chẩn đoán hội chứng Trichorhinophalangeal loại II có thể được thực hiện khi mới sinh bằng cách đánh giá biểu hiện lâm sàng và xác định các dấu hiệu đặc trưng. Chậm phát triển trí tuệ, mất thính giác hoặc chậm nói có thể không được phát hiện cho đến khi trẻ lớn hơn. Các xét nghiệm hình ảnh hỗ trợ phát hiện dị tật xương, u xương sụn. Tuy nhiên các triệu chứng và hình ảnh lâm sàng dễ gây nhầm lẫn với nhữung hội chứng tương tự. Xét nghiệm di truyền nhằm phát hiện các đột của gen TRPS1, EXT1 hoặc RAD21 có thể giúp chẩn đoán và đưa ra hướng điều trị phù hợp với từng bệnh nhân.
Điều trị
Hiện nay chưa có phương pháp điều trị hoàn toàn hội chứng Trichorhinophalangeal loại II. Các liệu pháp hướng đến triệu chứng cụ thể của mỗi người bệnh.
Một số trường hợp có thể chỉ định phẫu thuật cắt bỏ các u xương sụn gây đau, chèn ép dây thần kinh, cản trở quá trình phát triển của các chi hoặc gây suy giảm cử động. Vật lý trị liệu giúp cải thiện khả năng vận động. Máy trợ thính có thể được sử dụng điều trị chứng mất thính lực. Bệnh nhân cần được theo dõi thường xuyên và thực hiện các biện pháp phòng ngừa tránh nhiễm trùng đường hô hấp lặp lại.
Dạng di truyền
Bệnh chủ yếu xảy ra ngẫu nhiên trong quá trình hình thành các tế bào sinh sản (trứng hoặc tinh trùng) ở cha hoặc mẹ của trẻ bị bệnh. Những trường hợp này không có tiền sử bệnh trong gia đình của họ. Một vài trường hợp, người bệnh thừa hưởng đột biến mất đoạn nhiễm sắc thể từ cha hoặc mẹ bị bệnh.
Người ta cho rằng hội chứng Trichorhinophalangeal loại II di truyền theo kiểu trội trên nhiễm sắc thể thường. Theo những cáo cáo lâm sàng, người bệnh mang đột biến trên một bản sao của nhiễm sắc thể số 8 bị đứt gãy trong mỗi tế.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Để bảo đảm khả năng con không bị bệnh, cha mẹ có thể chọn phương pháp thụ tinh nhân tạo IVF và sàng lọc phôi PGS/PGD. Các thành viên trong gia đình nên khám sức khỏe và tầm soát bệnh định kỳ nếu có thành viên mắc bệnh. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh.
Các tên gọi khác
- Chromosome 8q24.1 deletion syndrome
- Giedion-Langer syndrome
- Langer-Giedion syndrome
- LGS
- Tricho-rhino-phalangeal syndrome type II
- Trichorhinophalangeal syndrome with exostosis
- TRPS II
- TRPS2
References
- Genetic Testing Information. Langer-Giedion syndrome. Retrieved October 14, 2022 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0023003/
- Genetic and Rare Diseases Information Center. Trichorhinophalangeal syndrome type 2. Retrieved October 14, 2022 from https://rarediseases.info.nih.gov/diseases/7801/trichorhinophalangeal-syndrome-type-2/
- Catalog of Genes and Diseases from OMIM. TRICHORHINOPHALANGEAL SYNDROME, TYPE II. Retrieved October 14, 2022 from https://omim.org/entry/150230
- U.S National Library of Medicine. Trichorhinophalangeal syndrome type II. Retrieved October 14, 2022 from https://medlineplus.gov/genetics/condition/trichorhinophalangeal-syndrome-type-ii/
- National Organization for Rare Disorders. Trichorhinophalangeal Syndrome Type II. Retrieved October 14, 2022 from https://rarediseases.org/rare-diseases/trichorhinophalangeal-syndrome-type-ii/
- MalaCards. Trichorhinophalangeal Syndrome, Type Ii (TRPS2). Retrieved October 14, 2022 from https://www.malacards.org/card/trichorhinophalangeal_syndrome_type_ii_2
- Orphanet. Trichorhinophalangeal syndrome type 2. Retrieved October 14, 2022 from https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=502
- National Foundation for Ectodermal Dysplasisa. TRICHORHINOPHALANGEAL SYNDROME, TYPE 2. Retrieved October 14, 2022 from https://www.nfed.org/learn/types/trichorhinophalangeal-syndrome-2/
- Hilton MJ, Sawyer JM, Gutiérrez L, Hogart A, Kung TC, Wells DE. Analysis of novel and recurrent mutations responsible for the tricho-rhino-phalangeal syndromes. Published 2002 https://doi.org/10.1007/s100380200010
- Schinzel A, Riegel M, Baumer A, Superti-Furga A, Moreira LM, Santo LD, Schiper PP, Carvalho JH, Giedion A. Long-term follow-up of four patients with Langer-Giedion syndrome: clinical course and complications. Published September, 2013 https://doi.org/10.1002/ajmg.a.36062





