Bệnh dày móng bẩm sinh (pachyonychia congenita) ảnh hưởng chủ yếu đến da và móng. Các đặc điểm của bệnh thường biểu hiện rõ ràng trong vài năm đầu đời.
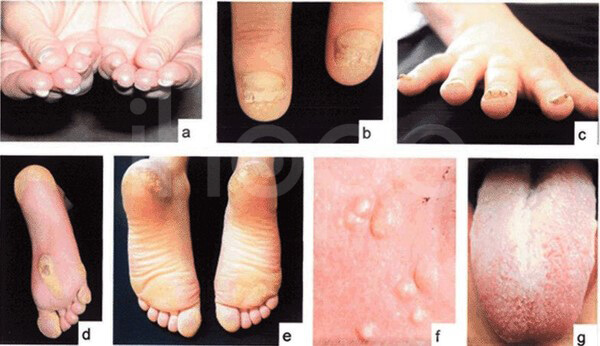
Ảnh: Đặc điểm của người mắc bệnh dày móng bẩm sinh
Nguồn: a-c) chứng loạn dưỡng móng phì đại, d-e) dày sừng lòng bàn tay-bàn chân, f) u nang, g) bệnh bạch cầu miệng
Nguồn: GeneReviews, © 1993-2020 University of Washington
Biểu hiện lâm sàng
Hầu hết người bệnh biểu hiện triệu chứng loạn dưỡng móng phì đại, họ có móng dày và hình dạng bất thường. Hầu hết trẻ mắc bệnh phát triển các vết phồng rộp và vết chai gây đau trong lòng bàn chân. Do đó, trẻ đi lại khó khăn hoặc không thể đi. Những triệu chứng này ít xuất hiện trong lòng bàn tay.
Ngoài ra, bệnh còn có một số triệu chứng không đồng nhất như:
- Các mảng trắng, dày trên lưỡi và bên trong má
- Dày sừng nang lông khuỷu tay, đầu gối và vùng eo
- U nang vùng nách, háng, lưng, da đầu
- Tăng tiết mồ hôi lòng bàn tay, bàn chân
- Trẻ mọc răng trước khi sinh
- Khàn tiếng hoặc khó thở
Nguồn: Terese Winslow
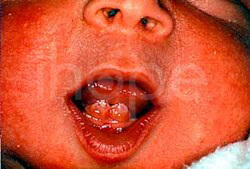
Ảnh: Trẻ mọc răng trước khi sinh
Nguồn: National Human Genome Research Institute
Độ phổ biến
Bệnh dày móng bẩm sinh hiếm gặp, do đó hiện nay tỷ lệ mắc bệnh vẫn chưa được thống kê cụ thể.
Nguyên nhân
Đột biến một số gen, bao gồm KRT6A, KRT6B, KRT6C, KRT16, KRT17 gây ra bệnh dày móng bẩm sinh. Các gen này cung cấp hướng dẫn tạo ra keratin - protein dạng sợi cứng tạo thành mạng lưới cung cấp độ bền và khả năng phục hồi cho mô da, tóc và móng tay.
Bệnh được phân loại dựa vào nguyên nhân di truyền, bao gồm:
- Đột biến gen KRT6A gây ra bệnh loại PC-K6a
- Đột biến gen KRT6B gây ra bệnh loại PC-K6b
- Đột biến gen KRT6C gây ra bệnh loại PC-K6c
- Đột biến gen KRT16 gây ra bệnh loại PC-K16
- Đột biến gen KRT17 gây ra bệnh loại PC-K17
Đột biến làm thay đổi cấu trúc của protein keratin, dẫn đến mạng lưới protein mất tính bền vững và kém ổn định trong tế bào. Do đó, tế bào da trở nên mỏng, giảm khả năng chống lại ma sát và dễ bị tổn thương. Những hoạt động bình thường như đi bộ cũng khiến các tế bào da bị phá vỡ rồi hình thành vết phồng rộp và chai nghiêm trọng gây đau đớn. Các chất sừng bất thường làm gián đoạn quá trình phát triển và hoạt động của tế bào trong nang tóc và móng tay, dẫn đến các đặc điểm khác của bệnh.
Chẩn đoán
Bác sĩ thường chẩn đoán bệnh dày móng bẩm sinh bằng các phương pháp sau:
- Kiểm tra thể chất, bao gồm da và móng tay
- Hỏi tiền sử bệnh của cá nhân và gia đình
- Hỏi các loại thuốc đã và đang sử dụng
- Thực hiện xét nghiệm di truyền tìm đột biến gen gây bệnh
Điều trị
Hiện nay chưa có phương pháp chữa khỏi bệnh dày móng bẩm sinh. Mục đích chính nhằm giảm đau và cải thiện chất lượng cuộc sống của người bệnh.
Các phương pháp điều trị vết chai, bao gồm:
- Làm mỏng vết chai bằng cách cắt da hoặc mài mòn
- Kem dưỡng ẩm
- Dùng thiết bị hỗ trợ như lót giày đặc biệt, gậy, nạng, xe lăn
- Retinoid đường uống
- Thuốc chống viêm
Các phương pháp điều trị móng và u nang, bao gồm:
- Cắt móng thường xuyên
- Dùng dung dịch rửa vệ sinh móng ngăn ngừa nhiễm trùng
- Thuốc kháng sinh hoặc thuốc kháng nấm đường uống
- Phẫu thuật cắt bỏ u nang
Nếu xuất hiện các mảng trắng trong miệng, người bệnh nên đánh răng và lưỡi nhẹ nhàng thường xuyên.
Dạng di truyền
Bệnh dày móng bẩm sinh di truyền theo kiểu trội trên nhiễm sắc thể thường. Một bản sao của gen bị thay đổi trong mỗi tế bào đủ để gây ra bệnh. Khoảng 60-70% người mắc bệnh thừa hưởng gen đột biến từ cha hoặc mẹ mang bệnh.

Nguồn: U.S. National Library of Medicine
Những trường hợp còn lại do một đột biến mới (de novo) trong gen xảy ra trong quá trình hình thành tế bào sinh sản (trứng hoặc tinh trùng) hoặc quá trình phát triển phôi sớm. Người bệnh không có tiền sử mắc bệnh trong gia đình của họ.
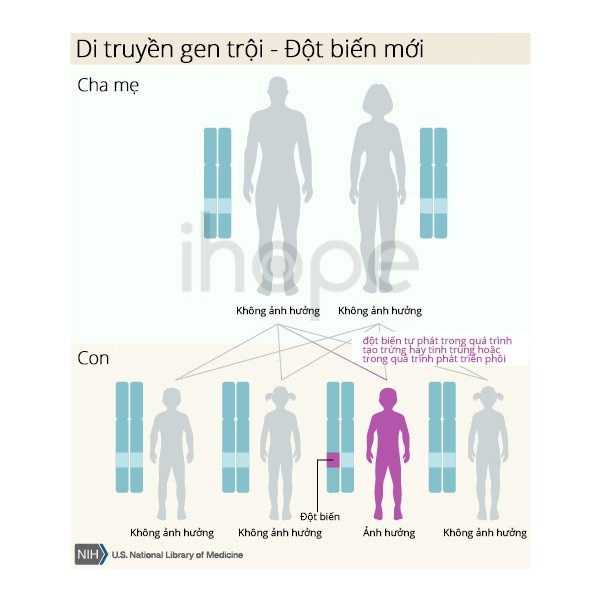
Nguồn: U.S. National Library of Medicine
Phòng ngừa
Bệnh di truyền trội trên nhiễm sắc thể thường, nếu cha hoặc mẹ mắc bệnh, con sinh ra sẽ có 50% khả năng di truyền bệnh. Do đó, để bảo đảm 100% khả năng con không bị bệnh, cha mẹ có thể chọn phương pháp thụ tinh nhân tạo IVF và sàng lọc phôi PGS/PGD. Các thành viên trong gia đình nên khám sức khỏe và tầm soát bệnh định kỳ nếu có thành viên mắc bệnh. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh.
Các tên gọi khác
- Congenital pachyonychia
- Jackson-Lawler syndrome (PC-2)
- Jadassohn-Lewandowski syndrome (PC-1)
- Pachyonychia congenita syndrome
References
- Genetic Testing Information. Pachyonychia congenita syndrome. Retrieved January 13, 2023 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0265334/
- Genetic and Rare Diseases Information Center. Pachyonychia congenita. Retrieved January 13, 2023 from https://rarediseases.info.nih.gov/diseases/10753/pachyonychia-congenita/
- Catalog of Genes and Diseases from OMIM. PACHYONYCHIA CONGENITA 1. Retrieved January 13, 2023 from https://omim.org/entry/167200
- U.S National Library of Medicine. Pachyonychia congenita. Retrieved January 13, 2023 from https://medlineplus.gov/genetics/condition/pachyonychia-congenita/
- National Institute of Arthritis and Musculoskeletal and Skin Diseases. Pachyonychia congenita. Retrieved January 13, 2023 from https://www.niams.nih.gov/health-topics/pachyonychia-congenita/diagnosis-treatment-and-steps-to-take
- Orphanet. Pachyonychia congenita. Retrieved January 13, 2023 from https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Expert=2309
- DermNet. Pachyonychia congenita. Retrieved January 13, 2023 from https://dermnetnz.org/topics/pachyonychia-congenita
- Eliason MJ, Leachman SA, Feng BJ, Schwartz ME, Hansen CD. A review of the clinical phenotype of 254 patients with genetically confirmed pachyonychia congenita. J Am Acad Dermatol. 2012 Oct;67(4):680-6. doi: 10.1016/j.jaad.2011.12.009.
- Wilson NJ, O'Toole EA, Milstone LM, Hansen CD, Shepherd AA, Al-Asadi E, Schwartz ME, McLean WH, Sprecher E, Smith FJ. The molecular genetic analysis of the expanding pachyonychia congenita case collection. Br J Dermatol. 2014 Aug;171(2):343-55. doi: 10.1111/bjd.12958.
- Smith FJ, Liao H, Cassidy AJ, Stewart A, Hamill KJ, Wood P, Joval I, van Steensel MA, Bjorck E, Callif-Daley F, Pals G, Collins P, Leachman SA, Munro CS, McLean WH. The genetic basis of pachyonychia congenita. J Investig Dermatol Symp Proc. 2005 Oct;10(1):21-30. doi: 10.1111/j.1087-0024.2005.10204.x.