Loạn sản hành xương sọ (craniometaphyseal dysplasia) là bệnh di truyền khiến xương sọ phát triển quá mức và gây ra các bất thường tại một vùng cuối xương dài được gọi là hành xương. Xương phát triển xương bất thường trong suốt cuộc đời bệnh nhân. Ngoại trừ các trường hợp nặng, tuổi thọ của bệnh nhân mắc loạn sản hành xương sọ vẫn bình thường.
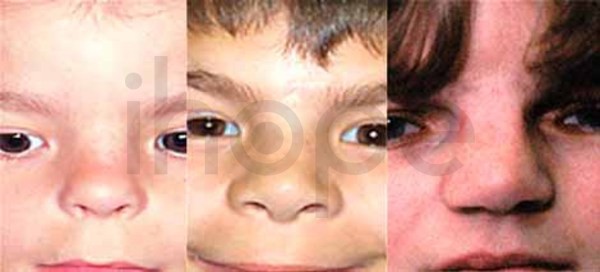
Ảnh: Mũi thấp và to
Nguồn: Elements of Morphology, National Human Genome Research Institute
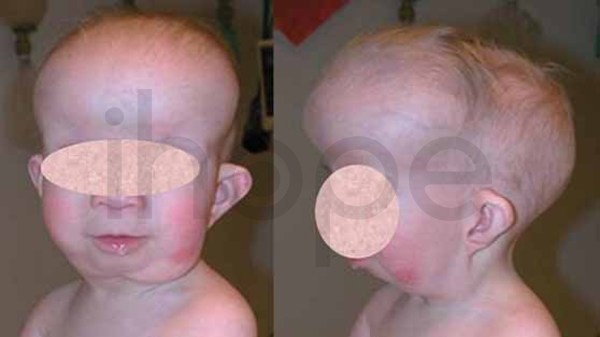
Ảnh: Trán nhô ra phía trước
Nguồn: Elements of Morphology, National Human Genome Research Institute
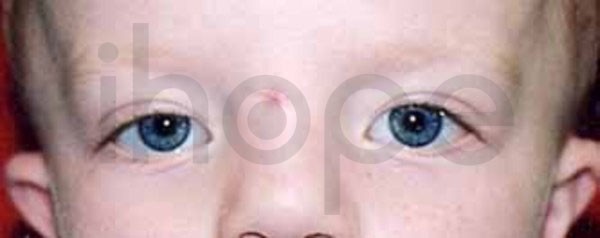
Ảnh: Khoảng cách giữa hai mắt rộng
Nguồn: Elements of Morphology, National Human Genome Research Institute
Biểu hiện lâm sàng
Xương hộp sọ phát triển quá mức trong gây ra nhiều dấu hiệu và triệu chứng của bệnh. Bệnh nhân thường có các đặc điểm khuôn mặt dị biệt bao gồm sống mũi rộng , trán dô , hai mắt xa nhau và hàm dưới nhô ra. Xương thừa trong hàm có thể làm chậm mọc răng hoặc khiến răng không mọc. Trẻ sơ sinh mắc bệnh có thể gặp các vấn đề hô hấp hoặc ăn uống do đường mũi hẹp. Trong các trường hợp nặng, xương phát triển bất thường có thể chèn ép các dây thần kinh sọ, từ đó dẫn đến liệt cơ mặt, mù lòa hoặc điếc.
Hình ảnh X-quang của bệnh nhân cho thấy các xương dài có hình dạng bất thường, chủ yếu là xương chân. Đầu của các xương này rộng hơn và có vẻ xốp hơn so với bình thường.
Độ phổ biến
Loạn sản hành xương sọ là bệnh di truyền hiếm gặp. Hiện nay, người ta chưa xác định được tỉ lệ mắc bệnh chính xác.
Nguyên nhân
Loạn sản hành xương sọ bắt nguồn từ đột biến gen ANKH di truyền trội trên nhiễm sắc thể thường. Gen ANKH cung cấp hướng dẫn tạo ra protein có vai trò trong quá trình phát triển và chức năng của các tế bào tạo xương (nguyên bào xương) và các tế bào phá hủy xương (hủy cốt bào). Hủy cốt bào tham gia vào quá trình tái tạo xương—quá trình sinh lí bình thường trong đó xương cũ được loại bỏ và xương mới được tạo ra để thay thế. Ngoài ra, protein ANKH vận chuyển phân tử pyrophosphate ra khỏi tế bào. Pyrophosphate ngoại bào có chức năng kiểm soát sự hình thành xương bằng cách ngăn chặn quá trình khoáng hóa. Đột biến gen ANKH làm suy giảm sự biệt hóa của hủy cốt bào, có khả năng phá vỡ quá trình tái tạo xương, từ đó góp phần vào sự dày lên của xương.
Đột biến gen GJA1 gây ra một số trường hợp loạn sản hành xương sọ dạng di truyền lặn trên nhiễm sắc thể thường. Gen này cung cấp hướng dẫn để tạo ra protein connexin 43—protein tham gia vào sự phát triển của nhiều mô trong cơ thể, bao gồm cả xương. Nguyên nhân di truyền của nhiều trường hợp di truyền lặn vẫn chưa rõ ràng.
Chẩn đoán
Chẩn đoán loạn sản hành xương sọ được thực hiện thông qua thăm khám bởi bác sĩ có chuyên môn về các rối loạn sọ mặt. Các phương pháp chẩn đoán hình ảnh như X-quang thường được yêu cầu nhằm chẩn đoán và theo dõi bệnh. Xét nghiệm di truyền có thể xác nhận chẩn đoán.
Điều trị
Hiện nay, chưa có phương pháp điều trị đặc hiệu cho loạn sản hành xương sọ. Các triệu chứng riêng lẻ được điều trị khi phát sinh. Quá trình chăm sóc bệnh nhân có thể bao gồm sự phối hợp của nhiều chuyên khoa.
Bệnh nhân mắc loạn sản hành xương sọ nên được can thiệp phẫu thuật sớm nhằm giải tỏa áp lực trong hộp sọ và thay đổi hình dạng xương mặt có thể giúp giảm một số biến chứng.
Dạng di truyền
Bệnh có hai dạng di truyền được phân biệt bởi kiểu di truyền và nguyên nhân di truyền.
Dạng trội trên nhiễm sắc thể thường được gây ra bởi đột biến gen ANKH, có nghĩa là một bản sao thay đổi của gen trong mỗi tế bào là đủ để gây ra rối loạn.

Nguồn: U.S. National Library of Medicine
Dạng lặn trên nhiễm sắc thể thường xảy ra do đột biến gen GJA1, nghĩa là cả hai bản sao của gen trong mỗi tế bào đều mang đột biến. Cha mẹ của một cá nhân mắc bệnh lặn trên nhiễm sắc thể thường mỗi người mang một bản sao của gen bị đột biến, nhưng họ thường không có dấu hiệu và triệu chứng của bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Tư vấn và xét nghiệm di truyền trước khi mang thai là cần thiết để đảm bảo sinh con khỏe mạnh. Đối với dạng di truyền trội, khi cha hoặc mẹ mắc bệnh, con sinh ra có 50% nguy cơ di truyền bệnh. Vì vậy, các phương pháp như thụ tinh trong ống nghiệm (IVF) kết hợp sàng lọc phôi có thể được cân nhắc. Đối với dạng di truyền lặn, cha mẹ mang gen đột biến thường không có biểu hiện, do đó xét nghiệm sàng lọc gen lặn được khuyến nghị để chủ động phòng ngừa. Các thành viên trong gia đình có người mắc bệnh nên thực hiện tầm soát di truyền và khám sức khỏe định kì.
Các tên gọi khác
- Autosomal dominant craniometaphyseal dysplasia
- Autosomal recessive craniometaphyseal dysplasia
- CMD
- CMDD
- CMDJ
- CMDR
- Craniometaphyseal dysplasia, Jackson type
References
- National Library of Medicine. Craniometaphyseal dysplasia, autosomal dominant. Retrieved September 21, 2025 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C1852502/
- National Organization for Rare Disorders. (2025). Craniometaphyseal Dysplasia. Retrieved September 21, 2025 from https://rarediseases.org/rare-diseases/craniometaphyseal-dysplasia/
- OMIM. CRANIOMETAPHYSEAL DYSPLASIA, AUTOSOMAL DOMINANT; CMDD. Retrieved September 21, 2025 from https://omim.org/entry/123000
- MedlinePlus. (2018). Craniometaphyseal dysplasia. Retrieved September 21, 2025 from https://medlineplus.gov/genetics/condition/craniometaphyseal-dysplasia/