Rối loạn glycosyl hóa bẩm sinh PMM2 (PMM2-congenital disorder of glycosylation) là bệnh di truyền ảnh hưởng đến nhiều cơ quan. Bệnh thường khởi phát vào giai đoạn sơ sinh với triệu chứng lâm sàng có thể khác nhau ở mỗi người bệnh.
Biểu hiện lâm sàng
Giai đoạn thai nhi
Thai nhi mắc bệnh thường có dấu hiệu phù thai do chất lỏng tích tụ bất thường trong một số cơ quan. Đa số trường hợp thai lưu hoặc trẻ qua đời ngay sau sinh.
Giai đoạn sơ sinh và trẻ nhỏ
Trẻ mắc bệnh có một số triệu chứng như:
- Yếu cơ và liệt chi
- Núm vú thụt vào trong
- Phân bố chất béo trên cơ thể bất thường
- Mắt lác
- Khó tăng cân
- Tiểu não kém phát triển
Nguồn: U.S. National Library of Medicine
Trẻ có khuôn mặt dị biệt bao gồm trán cao, khuôn mặt hình tam giác, tai lớn và môi trên mỏng. Những xét nghiệm chuyên sâu cho thấy tăng chức năng gan, tràn dịch màng ngoài tim và rối loạn đông máu .
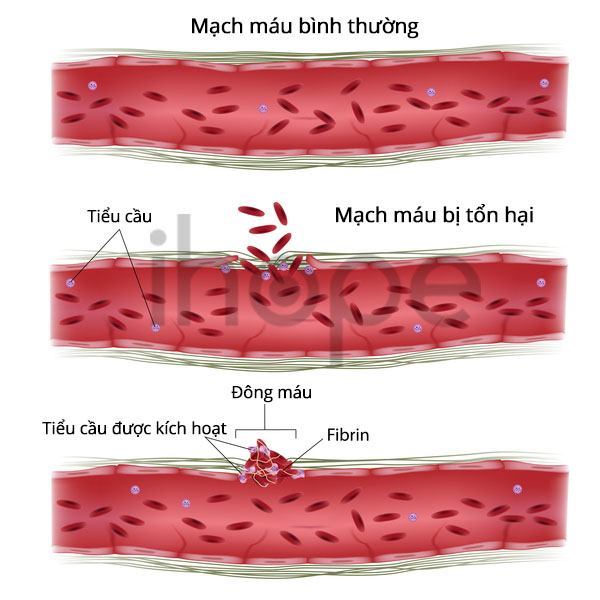
Ảnh: Cơ chế đông máu
Nguồn: Alila Medical Media/Shutterstock.com
Khoảng 20% trẻ mắc bệnh không sống sót qua 1 tuổi do suy đa cơ quan. Trẻ sống sót qua thời kỳ sơ sinh có thể bị thểu năng trí tuệ mức trung bình.
Giai đoạn thiếu niên và trưởng thành
Người bệnh có một số biểu hiện bao gồm:
- Cảm giác yếu cơ tay và chân
- Cong vẹo cột sống
- Mất điều hòa
- Biến dạng khớp
- Viêm võng mạc sắc tố
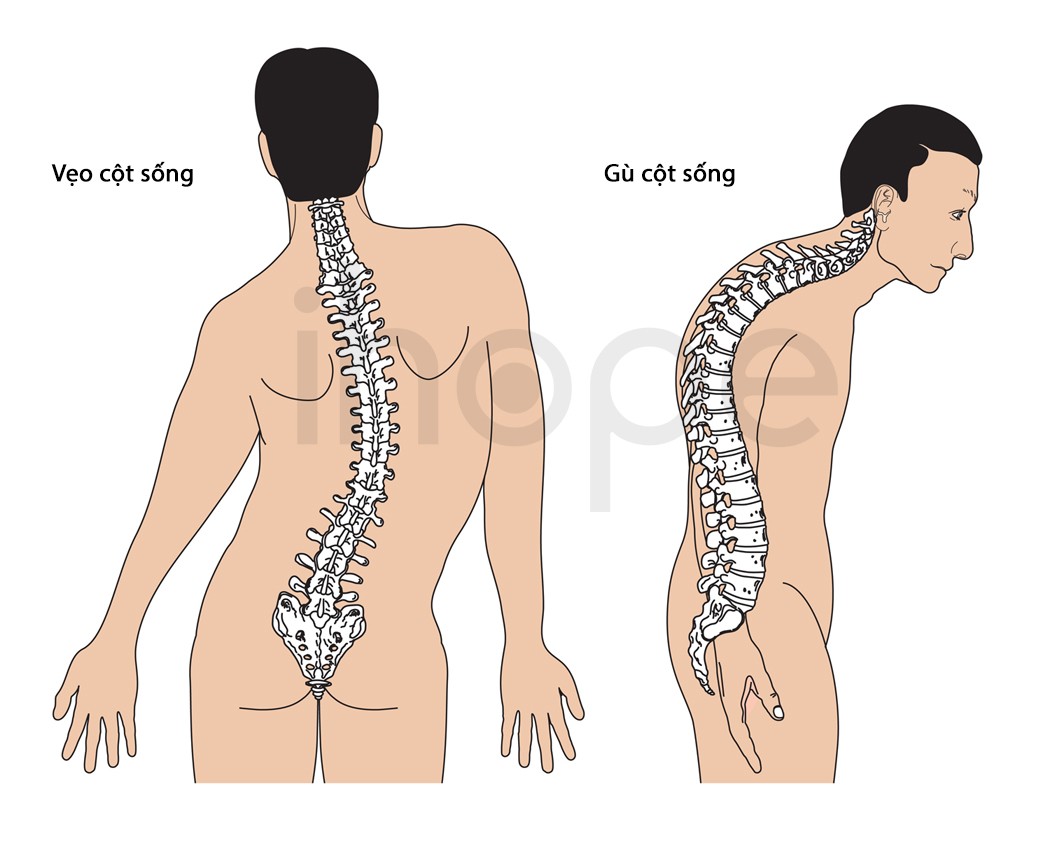
Ảnh: Độ cong bất thường của cột sống
Nguồn: Blamb/Shutterstock.com
Phụ nữ mắc bệnh phát triển chứng suy sinh dục, ảnh hưởng đến quá trình sản xuất hormone điều khiển quá trình phát triển giới tính. Do đó, họ không trải qua tuổi dậy thì. Nam giới bị bệnh trải qua tuổi dậy thì bình thường nhưng thường có tinh hoàn nhỏ.
Độ phổ biến
Hơn 800 trường hợp mắc bệnh được báo cáo trên toàn thế giới.
Nguyên nhân
Đột biến gen PMM2 gây ra chứng rối loạn glycosyl hóa bẩm sinh PMM2. Gen PMM2 cung cấp hướng dẫn tạo ra enzyme phosphomannomutase 2 (PMM2). Enzyme PMM2 tham gia vào quá trình glycosyl hóa, quá trình này gắn các nhóm phân tử đường (oligosaccharid) vào protein. Quá trình glycosyl hóa biến đổi protein để chúng thực hiện nhiều chức năng hơn.
Đột biến gen PMM2 dẫn đến sản xuất phiên bản enzyme PMM2 giảm hoạt tính nên quá trình glycosyl hóa không thể diễn ra bình thường. Các nhóm phân tử đường chưa hoàn chỉnh vẫn được gắn vào protein. Sự đa dạng của các triệu chứng trong PMM2-CDG có thể do quá trình sản xuất glycosyl hóa protein bất thường tại nhiều cơ quan và mô.
Chẩn đoán
Bệnh được chẩn đoán dựa trên triệu chứng, tiền sử bệnh nhân và một số xét nghiệm chuyên biệt để chẩn đoán.
Bác sĩ có thể chỉ định một số xét nghiệm như:
- Xét nghiệm máu phân tích nồng độ glycosyl hóa của transferrrin
- Xét nghiệm di truyền xác định đột biến gen PMM2
- Chụp cộng hưởng từ (MRI), đo điện não
- Kiểm tra tốc độ dẫn truyền thần kinh
Điều trị
Quy trình và biện pháp can thiệp điều trị tùy thuộc vào nhiều yếu tố bao gồm:
- Biểu hiện lâm sàng
- Mức độ nghiêm trọng
- Tuổi
- Sức khỏe tổng quát
- Khả năng đáp ứng với một số loại thuốc
Người bệnh cần được đảm bảo lượng calo nạp vào mỗi ngày. Một số trường hợp nặng có thể đặt ống thông qua một lỗ mở nhỏ trong dạ dày hoặc qua mũi.
Các đợt nhiễm trùng và sốt đột ngột làm tăng nguy cơ tử vong của trẻ mắc bệnh trong giai đoạn sơ sinh. Gia đình và bác sĩ cần theo dõi trẻ chặt chẽ nhằm có biến pháp điều trị kịp thời.
Dạng di truyền
Bệnh di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh lặn trên nhiễm sắc thể thường sẽ có bố và mẹ mang một bản sao của gen đột biến, nhưng bố mẹ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Bệnh di truyền lặn trên nhiễm sắc thể thường, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con.
Các thành viên trong gia đình nên khám sức khỏe và tầm soát bệnh định kỳ nếu có thành viên mắc bệnh. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh.
Các tên gọi khác
- Carbohydrate-deficient glycoprotein syndrome type Ia
- CDG Ia
- CDG1a
- CDGS1a
- Congenital disorder of glycosylation type Ia
- Jaeken syndrome
- Phosphomannomutase 2 deficiency
- PMM deficiency
- PMM2-CDG
References
- Genetic Testing Information. PMM2-congenital disorder of glycosylation. Retrieved December 19, 2022 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0349653/
- Genetic and Rare Diseases Information Center. PMM2-CDG (CDG-Ia). Retrieved December 19, 2022 from https://rarediseases.info.nih.gov/diseases/9826/pmm2-cdg-cdg-ia
- Catalog of Genes and Diseases from OMIM. CONGENITAL DISORDER OF GLYCOSYLATION, TYPE Ia. Retrieved December 19, 2022 from https://omim.org/entry/212065
- U.S National Library of Medicine. PMM2-congenital disorder of glycosylation. Retrieved December 19, 2022 from https://medlineplus.gov/genetics/condition/pmm2-congenital-disorder-of-glycosylation/
- National Organization for Rare Disorders. PMM2-CDG. Retrieved December 19, 2022 from https://rarediseases.org/rare-diseases/pmm2-cdg/
- Lam C, Krasnewich DM. PMM2-CDG. 2005 Aug 15 [updated 2021 May 20]. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2022. Available from http://www.ncbi.nlm.nih.gov/books/NBK1110/
- Orphanet. PMM2-CDG. Retrieved December 19, 2022 from https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=11344
- MalaCards. Congenital Disorder of Glycosylation, Type Ia (CDG1A). Retrieved December 19, 2022 from https://www.malacards.org/card/congenital_disorder_of_glycosylation_type_ia_2
- de Lonlay P, Seta N, Barrot S, Chabrol B, Drouin V, Gabriel BM, Journel H, Kretz M, Laurent J, Le Merrer M, Leroy A, Pedespan D, Sarda P, Villeneuve N, Schmitz J, van Schaftingen E, Matthijs G, Jaeken J, Korner C, Munnich A, Saudubray JM, Cormier-Daire V. A broad spectrum of clinical presentations in congenital disorders of glycosylation I: a series of 26 cases. J Med Genet. 2001 Jan;38(1):14-9. doi: 10.1136/jmg.38.1.14.
- Monin ML, Mignot C, De Lonlay P, Heron B, Masurel A, Mathieu-Dramard M, Lenaerts C, Thauvin C, Gerard M, Roze E, Jacquette A, Charles P, de Barace C, Drouin-Garraud V, Khau Van Kien P, Cormier-Daire V, Mayer M, Ogier H, Brice A, Seta N, Heron D. 29 French adult patients with PMM2-congenital disorder of glycosylation: outcome of the classical pediatric phenotype and depiction of a late-onset phenotype. Orphanet J Rare Dis. 2014 Dec 11;9:207. doi: 10.1186/s13023-014-0207-4.