Bệnh tích tụ lipofuscin neuron (neuronal ceroid lipofuscinose – NCL) hay bệnh Batten là nhóm bệnh ảnh hưởng đến hệ thần kinh, thị lực và trí tuệ. Bệnh được chia thành 8 loại dựa vào các đặc điểm sau:
- Nguyên nhân di truyền
- Độ tuổi khởi phát
- Dấu hiệu và triệu chứng
Tên của mỗi loại bao gồm “CLN” và số thứ tự loại phụ. Bệnh CLN5
Nguồn: U.S. National Library of Medicine
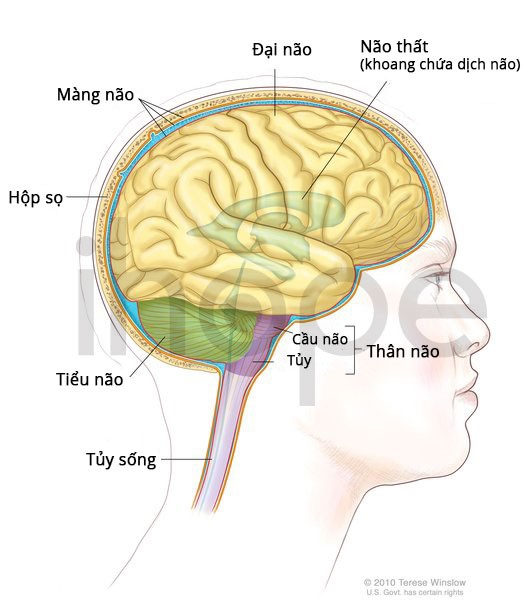
Biểu hiện lâm sàng
Bệnh có thể khởi phát từ thời thơ ấu đến đầu tuổi trưởng thành, nhưng đa số chúng thường xuất hiện vào khoảng 5 tuổi. Trẻ mắc bệnh phát triển bình thường cho đến khi xuất hiện các dấu hiệu đầu tiên, thường là các vấn đề vận động. Trẻ mất dần các kỹ năng đã học được trước đó như đi, đứng và cầm nắm.
Các đặc điểm khác của bệnh bao gồm:
- Động kinh
- Mất điều hòa
- Giảm thị lực
- Gặp vấn đề về giọng nói
- Thiểu năng trí tuệ
Người bệnh bị suy giảm tuổi thọ. Họ thường sống đến tuổi thiếu niên hoặc giữa tuổi trưởng thành.
Độ phổ biến
Bệnh CLN5 là bệnh di truyền hiếm gặp. Tỷ lệ mắc bệnh NCL trên toàn thế giới khoảng 1/100.00 người. Tại Phần Lan ước tính 1/12.500 người mắc bệnh NCL, bao gồm cả CLN5.
Nguyên nhân
Đột biến gen CLN5 gây ra bệnh CLN5. Gen CLN5 cung cấp hướng dẫn tạo ra một loại protein chưa rõ chức năng. Sau khi hình thành, protein CLN5 được vận chuyển đến lysosome - bộ phận tế bào đảm nhiệm tiêu hóa và tái chế nhiều loại phân tử khác nhau. Nghiên cứu cho thấy protein CLN5 tham gia vào quá trình phá vỡ lysosome và tái chế các protein bị hư hỏng hoặc không cần thiết trong tế bào.
Đa số đột biến gen CLN5 thay đổi cấu trúc của protein nên nó không thể di chuyển đến lysosome. Thiếu protein CLN5 trong lysosome làm suy yếu quá trình phân hủy một số protein. Protein dư thừa tích tụ đến mức gây hư hỏng các loại tế bào, trong đó tế bào thần kinh dễ bị tổn thương nhất. Mất tế bào thần kinh trên diện rộng gây ra các triệu chứng nghiêm trọng và đe dọa đến tính mạng.
Với những trường hợp bệnh phát triển vào tuổi thiếu niên hoặc trưởng thành, đột biến gen CLN5 dẫn đến một phiên bản protein CLN5 giảm chức năng và bị phân hủy sớm hơn bình thường. Protein CLN5 đã biến đổi hoạt động trong khoảng thời gian ngắn. Những protein hư hỏng hoặc không cần thiết có thể bị phân hủy trong lysosome. Chúng mất nhiều thời gian hơn để tích tụ và gây chết tế bào thần kinh. Do đó, bệnh biểu hiện triệu chứng trễ hơn.
Chẩn đoán
Người bệnh được chẩn đoán bệnh thông qua các biểu hiện lâm sàng và tiền sử bệnh trong gia đình. Bác sĩ có thể chỉ định một số xét nghiệm chuyên sâu hỗ trợ chẩn đoán.
Các xét nghiệm được thực hiện, bao gồm:
- Sinh thiết da đánh giá mức độ tích tụ mỡ
- Đo ERG kiểm tra phản ứng võng mạc với ánh sáng, các bất thường thần kinh thị giác
- Điện não đồ EEG
- Xét nghiệm enzyme loại trừ bệnh CLN1 và CLN2
- Xét nghiệm di truyền phát hiện gen đột biến gen CLN5
Điều trị
Hiện nay chưa có phương pháp điều trị hoàn toàn bệnh CLN5. Một số liệu pháp giúp giảm nhẹ triệu chứng bệnh như:
- Dùng thuốc
- Hỗ trợ thị lực
- Giáo dục đặc biệt kết hợp liệu pháp ngôn ngữ
Dạng di truyền
Bệnh di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh lặn trên nhiễm sắc thể thường sẽ có bố và mẹ mang một bản sao của gen đột biến, nhưng bố mẹ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Bệnh CLN5 di truyền lặn trên nhiễm sắc thể thường, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con.
Các thành viên trong gia đình nên khám sức khỏe và tầm soát bệnh định kỳ nếu có thành viên mắc bệnh. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh.
Các tên gọi khác
- Finnish variant late infantile neuronal ceroid lipofuscinosis
- Finnish vLINCL
- Jansky-Bielschowsky disease
- Late-infantile neuronal ceroid lipofuscinosis
- Neuronal ceroid lipofuscinosis 5
- Neuronal ceroid lipofuscinosis, late-infantile
- VLINCL
References
- Genetic Testing Information. Neuronal ceroid lipofuscinosis 5. Retrieved December 20, 2022 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C1850442/
- Genetic and Rare Diseases Information Center. Neuronal ceroid lipofuscinosis 5. Retrieved December 20, 2022 from https://rarediseases.info.nih.gov/diseases/1223/neuronal-ceroid-lipofuscinosis-5
- Catalog of Genes and Diseases from OMIM. CEROID LIPOFUSCINOSIS, NEURONAL, 5. Retrieved December 20, 2022 from https://omim.org/entry/256731
- U.S National Library of Medicine. CLN5 disease. Retrieved December 20, 2022 from https://medlineplus.gov/genetics/condition/cln5-disease/
- Orphanet. CLN5 disease. Retrieved December 20, 2022 from https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Expert=228360
- De Silva B, Adams J, Lee SY. Proteolytic processing of the neuronal ceroid lipofuscinosis related lysosomal protein CLN5. Exp Cell Res. 2015 Oct 15;338(1):45-53. doi: 10.1016/j.yexcr.2015.08.021.
- Kollmann K, Uusi-Rauva K, Scifo E, Tyynela J, Jalanko A, Braulke T. Cell biology and function of neuronal ceroid lipofuscinosis-related proteins. Biochim Biophys Acta. 2013 Nov;1832(11):1866-81. doi: 10.1016/j.bbadis.2013.01.019.
- Schmiedt ML, Bessa C, Heine C, Ribeiro MG, Jalanko A, Kyttala A. The neuronal ceroid lipofuscinosis protein CLN5: new insights into cellular maturation, transport, and consequences of mutations. Hum Mutat. 2010 Mar;31(3):356-65. doi: 10.1002/humu.21195.
- Williams RE, Mole SE. New nomenclature and classification scheme for the neuronal ceroid lipofuscinoses. Neurology. 2012 Jul 10;79(2):183-91. doi: 10.1212/WNL.0b013e31825f0547.
- Xin W, Mullen TE, Kiely R, Min J, Feng X, Cao Y, O'Malley L, Shen Y, Chu-Shore C, Mole SE, Goebel HH, Sims K. CLN5 mutations are frequent in juvenile and late-onset non-Finnish patients with NCL. Neurology. 2010 Feb 16;74(7):565-71. doi: 10.1212/WNL.0b013e3181cff70d.