
Mất thính giác không hội chứng (non-syndromic hearing loss) khiến người bệnh mất một phần hoặc toàn bộ khả năng nghe. Bệnh nhân thường không biểu hiện bất kì dấu hiệu hay triệu chứng nào. Mất thính giác không hội chứng di truyền theo nhiều cơ chế và mỗi trường hợp khởi phát bệnh tại từng thời điểm khác nhau.
Biểu hiện lâm sàng
Cấu trúc tai người gồm 3 phần chính: tai ngoài, tai giữa và tai trong. Phần lớn các dạng mất thính lực không triệu chứng do tổn thương tai trong. Tai trong có chức năng xử lí âm thanh và truyền thông tin đến não dưới dạng xung điện thần kinh. Bệnh do những thay đổi tại tai giữa ít phổ biến hơn. Tai giữa chứa ba xương nhỏ giúp truyền âm thanh từ màng nhĩ đến tai trong. Mất thính lực không triệu chứng dạng hỗn hợp ảnh hưởng cả tai trong và tai giữa của bệnh nhân.
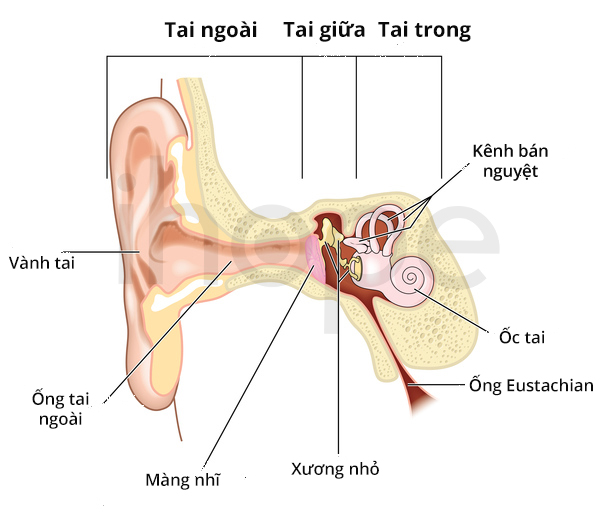
Nguồn: Blamb/Shutterstock.com
Người ta thường phân loại mất thính lực không triệu chứng dựa vào cơ chế di truyền bệnh. Các dạng bệnh bao gồm:
- Di truyền theo kiểu trội trên nhiễm sắc thể thường (DFNA)
- Di truyền theo kiểu lặn trên nhiễm sắc thể thường (DFNB)
- Di truyền theo kiểu lặn liên kết với nhiễm sắc thể giới tính X (DFNX)
- Di truyền theo ti thể (di truyền từ mẹ)
Mất thính lực không triệu chứng có thể khởi phát vào bất kì thời điểm nào từ giai đoạn sơ sinh đến tuổi già. Mỗi cá nhân có các dấu hiệu khác nhau, mất thính lực có thể ảnh hưởng một hoặc hai tai. Đối với mức độ nhẹ, người bệnh sẽ khó nghe được những âm thanh nhỏ. Trường hợp mất thính lực mức độ nặng, họ không thể nghe được, kể cả tiếng ồn lớn. Bệnh có thể ổn định hoặc tiến triển nặng hơn khi người bệnh lớn tuổi. Đối với một số dạng bệnh, biểu hiện mất thính lực xuất hiện rõ hơn khi người bệnh nghe âm thanh có tần số cao, trung bình hoặc thấp.
Độ phổ biến
Tại Mĩ, mỗi năm có khoảng 2–3/1.000 trẻ xuất hiện dấu hiệu mất thính lực tại một hoặc hai tai khi mới sinh. Nguy cơ mắc bệnh tăng dần theo độ tuổi. Cụ thể, tỉ lệ mắc bệnh đối với người từ 12 tuổi và từ 85 tuổi trở lên tại Mỹ lần lượt là 1/8 và 1/2 người.
Nguyên nhân
Người ta đã ghi nhận hơn 90 gen liên quan đến mất thính giác không hội chứng. Những gen này tham gia vào quá trình phát triển và hoạt động của tai trong. Các đột biến gen làm gián đoạn một số bước quan trọng trong quá trình xử lí âm thanh.
Đột biến di truyền theo kiểu lặn trên nhiễm sắc thể thường
Phần lớn các trường hợp mất thính giác không hội chứng di truyền theo kiểu lặn trên nhiễm sắc thể thường (bệnh DFNB). Trong đó, khoảng 50% trường hợp mất thính lực dạng nặng do đột biến gen GJB2. Gen này cung cấp hướng dẫn tạo ra protein connexin 26. Đột biến gen GJB6—gen cung cấp hướng dẫn tạo ra protein connexin 30 cũng gây ra bệnh.
Protein connexin tạo thành khe liên bào. Cấu trúc này liên kết các tế bào nhằm thúc đẩy những tế bào hiện diện tại tai trong tương tác với nhau. Đột biến gen GJB2 hoặc GJB6 thay đổi protein connexin, dẫn đến biến đổi cấu trúc khe liên bào. Do đó, chúng ảnh hưởng đến chức năng hoặc khả năng sống của những tế bào cần thiết cho thính giác.
Ngoài ra, đột biến gen STRC cũng gây ra bệnh DFNB. Gen STRC cung cấp hướng dẫn tạo ra protein stereocilin có chức năng liên kết đỉnh của các cấu trúc lông bất động tại tai trong. Lông bất động tham gia nhiều hoạt động liên quan đến chức năng thính giác như khuếch đại âm và phát hiện tần số âm thanh. Đột biến gen STRC làm mất chức năng của stereocilin, nên cấu trúc lông bất động bị ảnh hưởng theo, cuối cùng dẫn đến một số chức năng thính giác suy giảm.
Bên cạnh đó, đột biến tại hơn 60 gen khác cũng có thể gây mất thính lực không triệu chứng dạng DFNB. Người ta đã ghi nhận những trường hợp đột biến này trong một số gia đình.
Đột biến di truyền theo kiểu trội trên nhiễm sắc thể thường
Người ta đã ghi nhận ít nhất 30 đột biến gen liên quan đến mất thính giác không hội chứng di truyền theo kiểu trội trên nhiễm sắc thể thường (bệnh DFNA). Một số đột biến di truyền trội cũng có thể di truyền theo kiểu lặn (ví dụ như đột biến gen GJB2 và GJB6). Hai đột biến gen phổ biến nhất gây ra bệnh DFNA là KCNQ4 và TECTA.
Gen KCNQ4 cung cấp hướng dẫn tạo ra protein thuộc nhóm kênh vận chuyển ion kali hiện diện trên màng tế bào. Protein này có chức năng duy trì lượng ion kali phù hợp tại tai trong nhằm thúc đẩy quá trình truyền xung điện thần kinh từ tai trong đến não. Đột biến gen KCNQ4 có thể ngăn cản protein này di chuyển đến màng tế bào hoặc ảnh hưởng đến chức năng vận chuyển ion của chúng. Do đó, ion kali tích tụ trong một số tế bào tai trong đế mức làm tổn thương tế bào và gây ra mất thính lực.
Gen TECTA cung cấp hướng dẫn tạo ra protein alpha-tectorin hiện diện trên màng mái của ốc tai. Ốc tai có chức năng chuyển tín hiệu sóng âm thành xung thần kinh rồi truyền đến não. Một số đột biến TECTA ảnh hưởng đến khả năng nghe âm thanh tại tần số trung bình hoặc cao. Những đột biên khác thay đổi cấu trúc màng mái, do đó quá trình chuyển đổi âm thanh thành xung thần kinh bị cản trở.
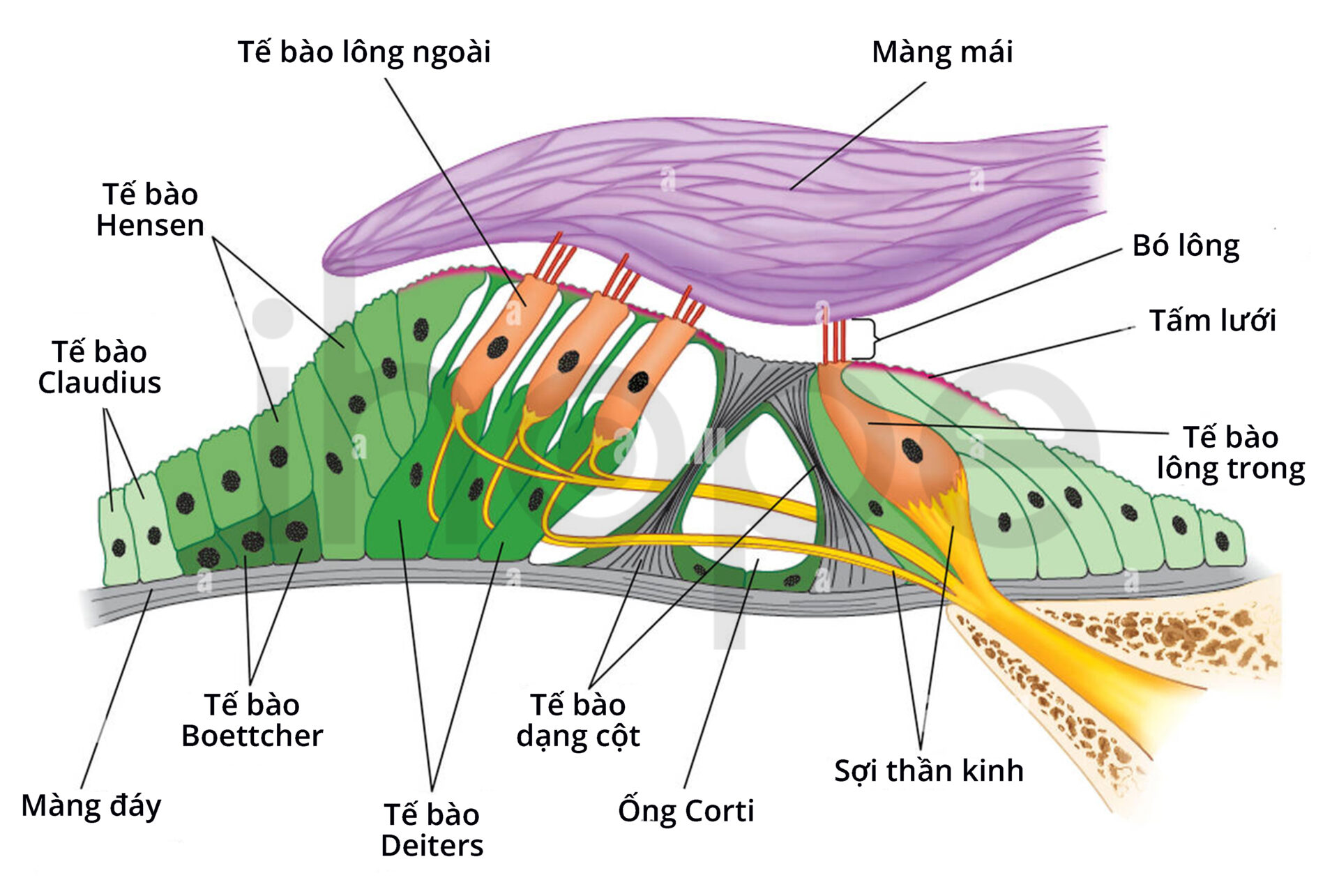
Nguồn: Encyclopædia Britannica, Inc.
Đột biến di truyền theo kiểu lặn liên kết với nhiễm sắc thể X và theo ti thể
Đột biến gen POU3F4 gây ra mất thính giác không hội chứng di truyền theo kiểu lặn liên kết với nhiễm sắc thể X (bệnh DFNX). Gen POU3F4 cung cấp hướng dẫn tạo ra protein POU3F4 thuộc nhóm yếu tố phiên mã họ POU. Những yếu tố này rất quan trọng đối với quá trình phát triển tai giữa, tai trong và một số vùng trong não trẻ trước khi sinh. Protein POU3F4 chứa một số vùng chuyên biệt giúp nó liên kết với các đoạn ADN .
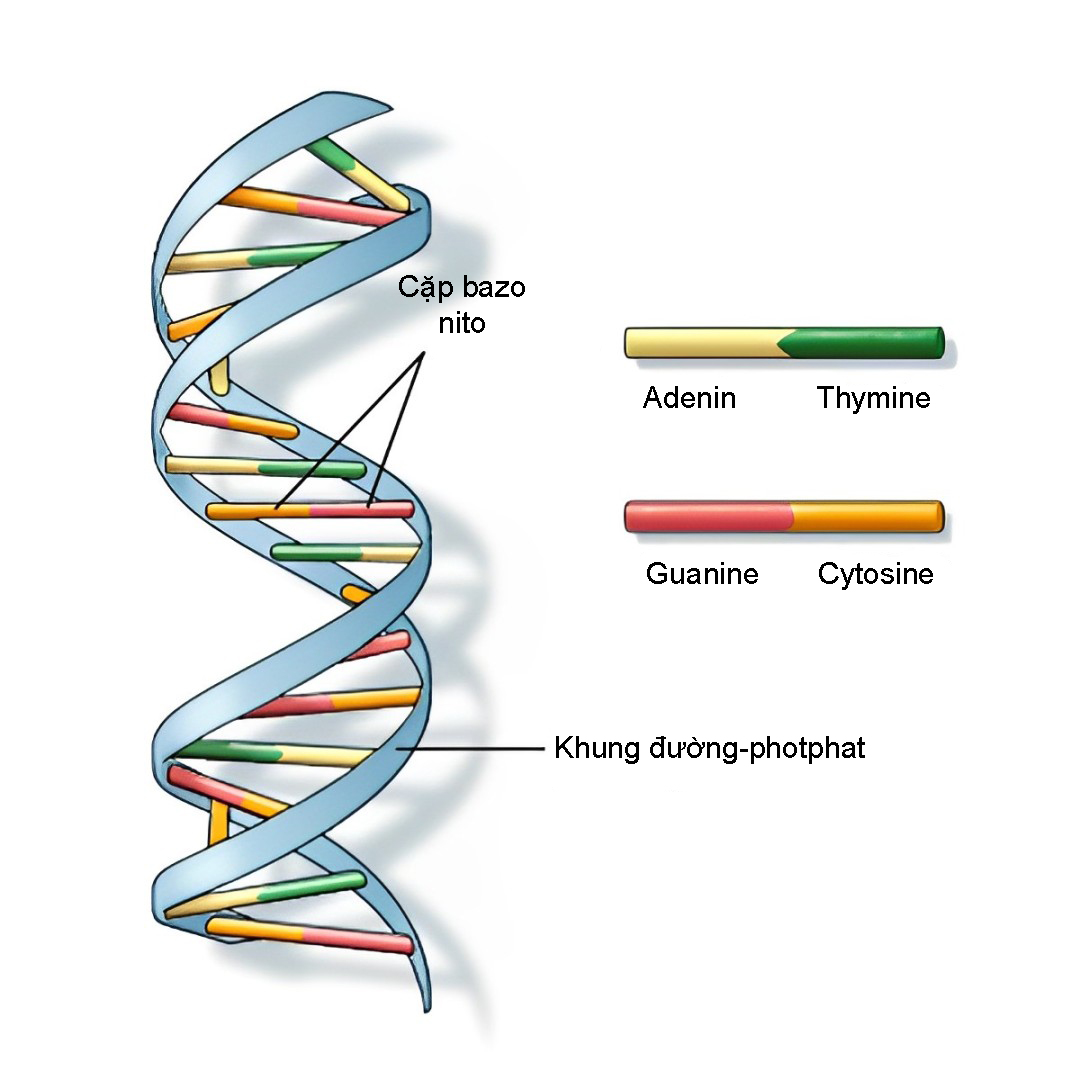
Ảnh: Cấu trúc của ADN
Nguồn: U.S. National Library of Medicine
Các đột biến gần hoặc trên gen POU3F4 có thể ngăn cản quá trình tạo ra protein POU3F4 hoặc làm tổn hại vùng liên kết với ADN. Do đó, quá trình phát triển bình thường tai giữa và tai trong bị gián đoạn, dẫn đến người bệnh bị mất thính lực. Người ta cũng ghi nhận một số đột biến trên ba gen khác liên quan đến bệnh DFNX.
Những biến đổi trong ADN ti thể gây ra mất thính giác không hội chứng di truyền theo ti thể. Ti thể là cấu trúc có chức năng chuyển đổi năng lượng từ thức ăn thành dạng năng lượng tế bào có thể sử dụng. Người ta mới ghi nhận một số đột biến ADN ti thể liên quan đến mất thính lực và đang tìm hiểu mối liên hệ giữa những đột biến này với bệnh.
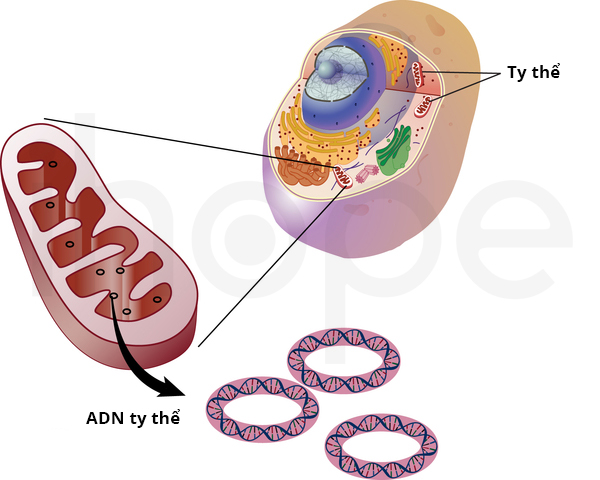
Nguồn: Darryl Leja, NHGRI
Đột biến gây mất thính lực có triệu chứng
Một số đột biến gen gây mất thính giác không hội chứng cũng có thể dẫn đến những dạng mất thính lực có triệu chứng như:
- CDH23 và MYO7A: hội chứng Usher
- SLC26A4: hội chứng Pendred
- WFS1: hội chứng Wolfram
- COL11A2: hội chứng Stickler
Hiện nay, người ta chưa rõ cơ chế gây bệnh của những đột biến này.
Các yếu tố môi trường
Những yếu tố sau có thể làm tăng nguy cơ mất thính lực:
- Sử dụng một số loại thuốc ảnh hưởng đến thính lực
- Nhiễm trùng tai
- Nghe âm thanh lớn trong thời gian dài
- Tuổi tác tăng
Chẩn đoán
Bác sĩ thường chẩn đoán mất thính giác không hội chứng dựa vào tiền sử bệnh gia đình, tiền sử thai kì cũng như kết quả khám sức khoẻ, đo thính lực và xét nghiệm di truyền.
Đối với trẻ sơ sinh, bác sĩ sàng lọc khả năng nghe của trẻ bằng cách đo mức độ phản hồi của tế bào lông ngoài hoặc trong ốc tai, dây thần kinh thính giác, thân não và não với các kích thích âm thanh.
Nếu kết quả sàng lọc cho thấy dấu hiệu bất thường, bác sĩ sẽ chỉ định trẻ thực hiện thêm các xét nghiệm đo thính lực khác nhằm chẩn đoán chuyên sâu. Bác sĩ tai mũi họng quan sát tai của trẻ nhằm đánh giá các nguyên nhân gây bệnh như viêm tai giữa, cấu trúc tai ngoài và tai giữa bất thường cũng như dấu hiệu của một số hội chứng khác liên quan đến mất thính lực.
Xét nghiệm di truyền đa gen giúp phát hiện các đột biến gen liên quan đến bệnh và gen gây ra biểu hiện tương tự bệnh. Ngoài ra, bác sĩ có thể chỉ định giải trình tự exome (tập hợp các trình tự ADN cung cấp hướng dẫn tạo ra protein) hoặc giải trình tự bộ gen.
Điều trị
Hiện nay, chưa có phương pháp điều trị hoàn toàn mất thính giác không hội chứng. Bác sĩ thường chỉ định ghép ốc tai điện tử hoặc sử dụng máy trợ thính nhằm cải thiện khả năng nghe của người bệnh.
Ốc tai điện tử
Ốc tai điện tử tạo ra đường truyền tín hiệu mới từ tai ngoài đến tai trong. Người bệnh đeo một loại thiết bị xử lí âm thanh. Thiết bị này tiếp nhận tín hiệu sóng âm và gửi đến bộ phận truyền được gắn trên da đầu. Sau đó, bộ phận truyền chuyển tín hiệu âm thanh thành xung điện và truyền đến điện cực đặt tại ốc tai. Điện cực thu nhận xung điện và truyền đến các dây thần kinh thính giác.

Nguồn: Cleveland Clinic
Máy trợ thính
Máy trợ thính bao gồm ba bộ phận chính:
- Micro
- Bộ khuếch đại
- Loa (máy thu)
Khi âm thanh đi qua micro, micro chuyển đổi sóng âm thành tín hiệu điện và gửi chúng đến bộ khuếch đại. Sau đó, bộ khuếch đại tăng cường độ tín hiệu điện, chuyển nó về dạng sóng âm và gửi đến tai người bệnh thông qua loa.
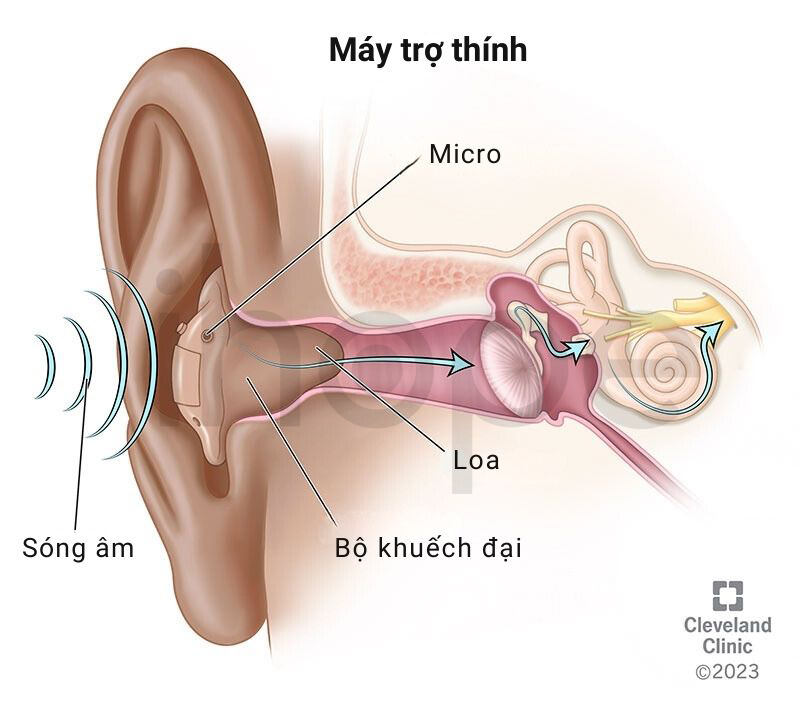
Nguồn: Cleveland Clinic
Dạng di truyền
Khoảng 75–80% trường hợp mất thính lực không triệu chứng di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh lặn trên nhiễm sắc thể thường sẽ có bố và mẹ mang một bản sao của gen đột biến, nhưng họ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Khoảng 20–25% trường hợp bệnh di truyền theo kiểu trội trên nhiễm sắc thể thường. Do đó, một bản sao của gen trong mỗi tế bào đủ để gây ra bệnh. Người bệnh thường được di truyền từ cha hoặc mẹ mắc bệnh. Một số ít trường hợp là do đột biến mới (denovo) xuất hiện trong quá trình hình thành tế bào sinh sản (trứng, tinh trùng) hoặc quá trình phát triển phôi sớm.

Nguồn: U.S. National Library of Medicine
Khoảng 1–2% trường hợp bệnh di truyền theo kiểu lặn liên kết với nhiễm sắc thể giới tính X. Nam giới chỉ có một nhiễm sắc thể X, do đó một bản sao của gen đột biến trong mỗi tế bào đủ gây ra triệu chứng nghiêm trọng bệnh. Người cha bị bệnh không thể truyền gen đột biến này cho con trai. Đối với phụ nữ có hai nhiễm sắc thể X, đột biến tại một bản sao của gen thường chỉ gây ra các triệu chứng nhẹ.
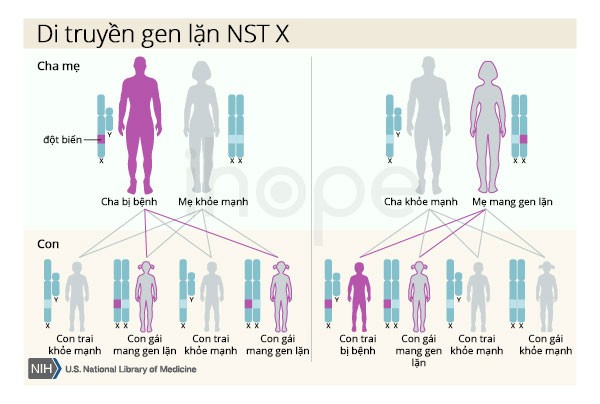
Nguồn: U.S. National Library of Medicine
Dạng di truyền theo ti thể hay còn gọi là di truyền từ mẹ chiếm ít hơn 1% trong số các trường hợp mất thính lực không triệu chứng. Vì phôi chỉ nhận ti thể từ tế bào trứng nên trẻ chỉ thừa hưởng những đột biến gen trong ti thể của người mẹ. Người cha mắc bệnh không thể truyền gen đột biến này cho con. Bệnh có thể ảnh hưởng cả nam và nữ.
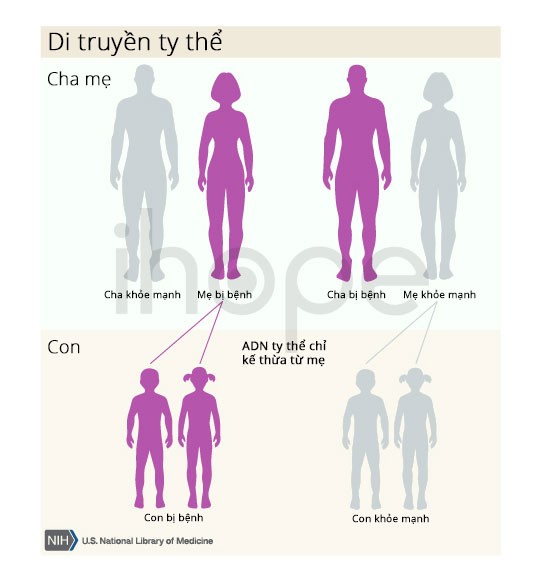
Nguồn: U.S. National Library of Medicine
Phòng ngừa
Mất thính giác không hội chứng di truyền lặn do nhiều đột biến gen, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con.
Trong trường hợp bệnh di truyền trội trên nhiễm sắc thể thường, nếu cha hoặc mẹ mắc bệnh, con sinh ra sẽ có 50% khả năng di truyền bệnh. Do đó, để bảo đảm 100% khả năng con không bị bệnh, cha mẹ có thể chọn phương pháp thụ tinh nhân tạo IVF và sàng lọc phôi PGS/PGD.
Bệnh có cơ chế di truyền liên kết X phức tạp nên khó phát hiện trên những phụ nữ mang gen bệnh cho đến khi sinh con. Để chủ động phòng ngừa, người mẹ nên làm xét nghiệm sàng lọc gen lặn.
Bệnh có thể di truyền theo ti thể, người mẹ mắc bệnh sẽ sinh ra con mắc bệnh. Do đó, phụ nữ trước khi mang thai cần làm xét nghiệm sàng lọc đột biến gen ti thể. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh hoặc có tiền sử bệnh trong gia đình cần khám sức khỏe và tầm soát bệnh định kì.
Các tên gọi khác
- Isolated deafness
- Nonsyndromic deafness
- Nonsyndromic hearing impairment
- Nonsyndromic hearing loss and deafness
References
- Genetic Testing Information. Nonsyndromic genetic hearing loss. Retrieved July 08, 2024 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C5680182/
- Genetic and Rare Diseases Information Center. Nonsyndromic hereditary sensorineural hearing loss. Retrieved July 08, 2024 from https://rarediseases.info.nih.gov/diseases/7222/index
- Catalog of Genes and Diseases from OMIM. DEAFNESS, AUTOSOMAL DOMINANT 7; DFNA7. Retrieved July 08, 2024 from https://omim.org/entry/601412
- U.S National Library of Medicine. Nonsyndromic hearing loss. Retrieved July 08, 2024 from https://medlineplus.gov/genetics/condition/nonsyndromic-hearing-loss/
- Centers for Disease Control and Prevention About the Types of Hearing Loss. Retrieved July 08, 2024 from https://www.cdc.gov/hearing-loss-children-guide/parents-guide-genetics/about-the-types-of-hearing-loss.html
- Frontiers. Recent advances in genetic etiology of non-syndromic deafness in children. Retrieved July 08, 2024 from https://www.frontiersin.org/journals/neuroscience/articles/10.3389/fnins.2023.1282663/full
- National Institute of Health. Genetic Hearing Loss Overview. Retrieved July 08, 2024 from https://www.ncbi.nlm.nih.gov/books/NBK1434/
- Orphanet. Non-syndromic genetic deafness. Retrieved July 08, 2024 from https://www.orpha.net/en/disease/detail/87884






