
Thiếu protein lưỡng-năng-D (D-bifunctional protein deficiency) là di truyền gây suy giảm chức năng hệ thần kinh (thoái hóa thần kinh) trên trẻ nhỏ. Trẻ mắc bệnh thường bị nhược cơ và co giật.
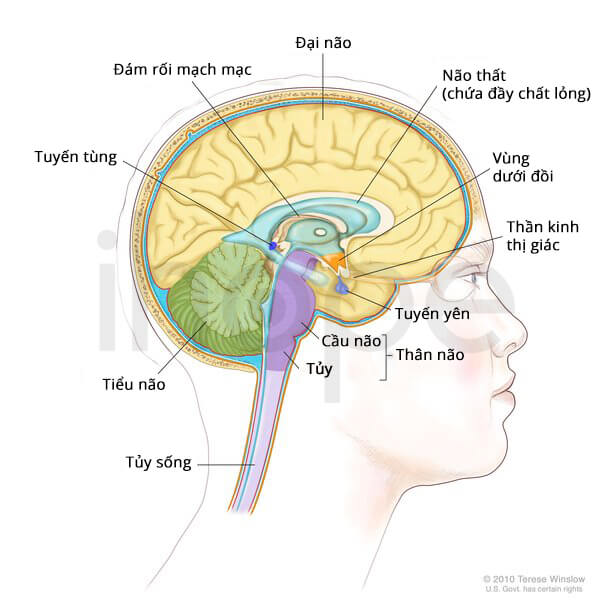
Nguồn: Terese Winslow
Biểu hiện lâm sàng
Phần lớn trẻ em thiếu protein lưỡng-năng-D không có bất kỳ kỹ năng phát triển nào. Một số trẻ có thể quan sát chuyển động bằng mắt hoặc kiểm soát cử động đầu, tuy nhiên, những kỹ năng này thường mất dần trong vòng vài tháng (thoái trào phát triển).
Khi bệnh tiến triển nghiêm trọng, bệnh nhân khởi phát một số triệu chứng bao gồm:
- Tăng phản xạ (hyperreflexia)
- Tăng trương lực cơ (hypertonia hyperreflexia)
- Co giật
- Động kinh
- Mất thị giác và thính giác
Đa số trẻ em thiếu protein lưỡng-năng-D sống không quá 2 năm. Một số người bệnh có triệu chứng ít nghiêm trọng có thể cử động tay theo ý muốn hoặc ngồi không cần hỗ trợ. Những bệnh nhân này có thể sống lâu hơn so với người mắc bệnh nặng.
Người bệnh có các bất thường trên khuôn mặt, bao gồm trán cao, hai mắt cách xa nhau , nhân trung dài, vòm cứng của khoang miệng cao bất thường. Trẻ sơ sinh có một khoảng trống lớn giữa các xương sọ. Khoảng một nửa số trường hợp mắc bệnh phát triển chứng gan to. Bởi vì giống với những đặc điểm của hội chứng Zellweger, thiếu protein lưỡng-năng-D đôi khi được gọi là hội chứng giả Zellweger.
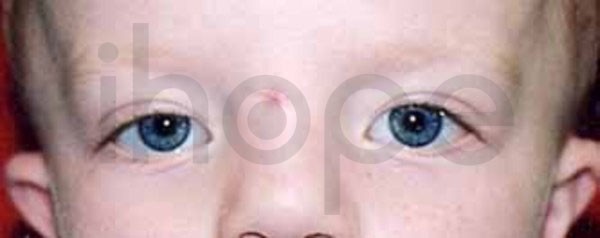
Ảnh: Khoảng cách giữ hai mắt rộng
Nguồn: Elements of Morphology, National Human Genome Research Institute
Độ phổ biến
Người ta ước tính tỷ lệ mắc bệnh thiếu protein lưỡng-năng-D khoảng 1/100.000 trẻ sơ sinh.
Nguyên nhân
Đột biến gen HSD17B4 gây ra chứng thiếu protein lưỡng-năng-D. Gen HSD17B4 cung cấp hướng dẫn tạo ra protein lưỡng-năng-D. Protein này hiện diện trong bào quan peroxisome—cấu trúc tế bào giống như túi chứa nhiều loại enzyme phân hủy nhiều chất khác nhau.
Đột biến gen HSD17B4 làm suy giảm số lượng protein dẫn đến quá trình phân hủy axit béo kém hiệu quả. Axit béo tích tụ trong cơ thể đến mức khiến não bộ phát triển bất thường. Ngoài ra, axit béo dư thừa phá vỡ bao myelin—thành phần có nhiệm vụ bảo vệ dây thần kinh và thúc đẩy quá trình dẫn truyền xung thần kinh. Do đó, mô chứa myelin (chất trắng) trong não và tủy sống bị mất đi (loạn dưỡng chất trắng), từ đó dẫn đến các triệu chứng của bệnh.
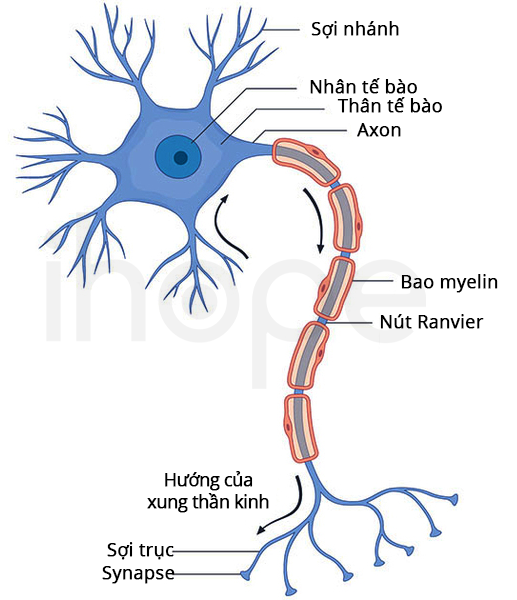
Nguồn: Eunice Kennedy Shriver National Institute of Child Health and Human Development
Chẩn đoán
Chẩn đoán thiếu protein lưỡng-năng-D dựa trên các biểu hiện lâm sàng và tiền sử bệnh trong gia đình. Ngoài ra, bác sĩ có thể thực hiện một số xét nghiệm chuyên biệt như đo nồng độ axit béo chuỗi rất dài tích tụ trong huyết tương. Chụp cộng hưởng từ (MRI) não có thể hỗ trợ chẩn đoán bệnh.
Điều trị
Hiện nay chưa có phương pháp điều trị hoàn toàn chứng thiếu protein lưỡng-năng-D. Các liệu pháp nhằm giảm nhẹ triệu chứng và cải thiện chất lượng đời sống bệnh nhân.
Dạng di truyền
Bệnh thiếu protein lưỡng-năng-D di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh lặn trên nhiễm sắc thể thường sẽ có bố và mẹ mang một bản sao của gen đột biến, nhưng bố mẹ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Bệnh thiếu protein lưỡng-năng-D di truyền lặn do đột biến gen HSD17B4, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kỳ.
Các tên gọi khác
- 17-beta-hydroxysteroid dehydrogenase IV deficiency
- Bifunctional peroxisomal enzyme deficiency
- DBP deficiency
- PBFE deficiency
- Peroxisomal bifunctional enzyme deficiency
- Pseudo-Zellweger syndrome
- Zellweger-like syndrome
References
- Genetic Testing Information. Bifunctional peroxisomal enzyme deficiency. Retrieved 24 August 2023 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0342870/?_ga=2.240417989.739754063.1692582707-1635318420.1684225451
- Genetic and Rare Diseases Information Center. Bifunctional enzyme deficiency. Retrieved 24 August 2023 from https://rarediseases.info.nih.gov/diseases/4539/d-bifunctional-protein-deficiency
- Catalog of Genes and Diseases from OMIM. D-BIFUNCTIONAL PROTEIN DEFICIENCY. Retrieved 24 August 2023 from https://omim.org/entry/261515
- U.S National Library of Medicine. D-bifunctional protein deficiency. Retrieved 24 August 2023 from https://medlineplus.gov/genetics/condition/d-bifunctional-protein-deficiency/
- Frontiers. Two Novel HSD17B4 Heterozygous Mutations in Association With D-Bifunctional Protein Deficiency: A Case Report and Literature Review. Retrieved 24 August 2023 from https://www.frontiersin.org/articles/10.3389/fped.2021.679597/full
- MalaCards. D-Bifunctional Protein Deficiency (DBPD). Retrieved 24 August 2023 from https://www.malacards.org/card/d_bifunctional_protein_deficiency
- MSD Manuals. Peroxisome Biogenesis and Very Long-Chain Fatty Acid Metabolism Disorders. Retrieved 24 August 2023 from https://www.msdmanuals.com/professional/multimedia/table/peroxisome-biogenesis-and-very-long-chain-fatty-acid-metabolism-disorders
- National Organization for Rare Disorders. D-bifunctional protein deficiency. Retrieved 24 August 2023 from https://rarediseases.org/gard-rare-disease/d-bifunctional-protein-deficiency/
- Orphanet. Bifunctional enzyme deficiency. Retrieved 24 August 2023 from https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=GB&Expert=300





