
Aspartylglucosamin niệu (aspartylglucosaminuria) là bệnh hiếm gặp do thiếu enzyme aspartylglycosaminidase trong lysosome . Người bệnh có triệu chứng tích tụ protein glycoasparagine tại các mô trong cơ thể và tăng bài tiết protein này qua nước tiểu.
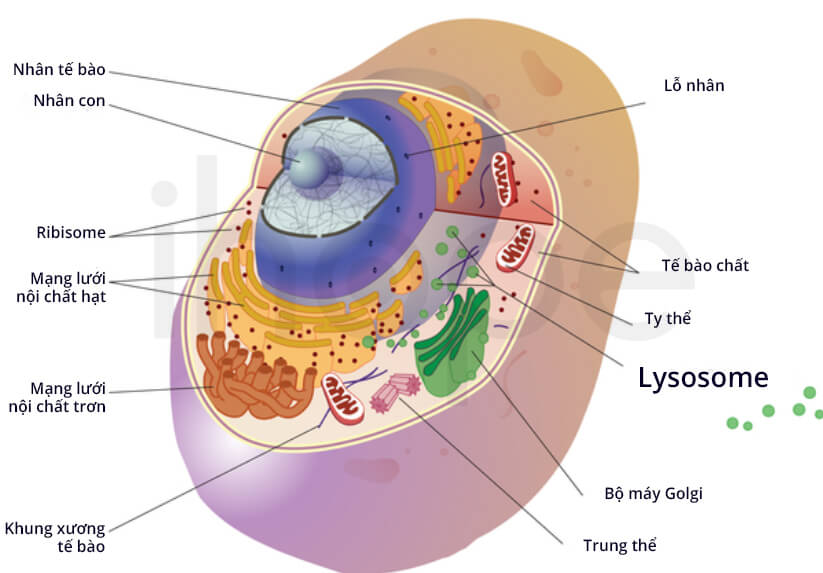
Ảnh: Lysosome và các bào quan khác
Nguồn: Darryl Leja, NHGRI
Biểu hiện lâm sàng
Aspartylglucosamin niệu chủ yếu ảnh hưởng đến chức năng thần kinh và vận động của người bệnh. Bệnh thường khởi phát từ độ tuổi 2–3 và dần trở nên nghiêm trọng hơn khi bệnh nhân trưởng thành.
Chức năng thần kinh
Bệnh nhi 2 hoặc 3 tuổi thường có biểu hiện:
- Chậm nói
- Thiểu năng trí tuệ dạng nhẹ
- Gặp vấn đề về phối hợp cử động
Phần lớn bệnh nhân mất khả năng nói đã học trước đó và có vốn từ vựng hạn chế. Bệnh nhân có thể gặp các rối loạn thần kinh như tăng động, lo âu, bồn chồn và thờ ơ. Ngoài ra, họ có khả năng rối loạn giấc ngủ, động kinh cũng như những bệnh lí liên quan đến chức năng vận động của cơ thể.
Đặc điểm khuôn mặt
Bệnh nhân thường có khuôn mặt dị biệt với các đặc điểm như:
- Kích thước mặt lớn, có dạng hình vuông
- Khoảng cách giữa hai mắt xa nhau
- Tai nhỏ
- Viền môi trên dày
- Lưỡi to
- Mũi ngắn với đầu mũi lớn
- Hàm dưới chìa ra
Trẻ em mắc bệnh thường phát triển nhanh dẫn đến đầu có kích thước lớn bất thường. Tuy nhiên, khi người bệnh đến tuổi dậy thì, tốc độ tăng trưởng sẽ chậm lại. Vì vậy, họ có dáng người thấp bé và kích thước đầu nhỏ khi trưởng thành.
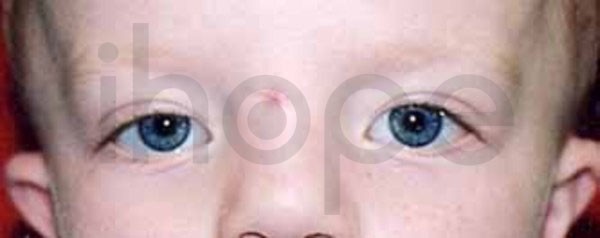
Ảnh: Khoảng cách giữa hai mắt xa nhau
Nguồn: Elements of Morphology, National Human Genome Research Institute
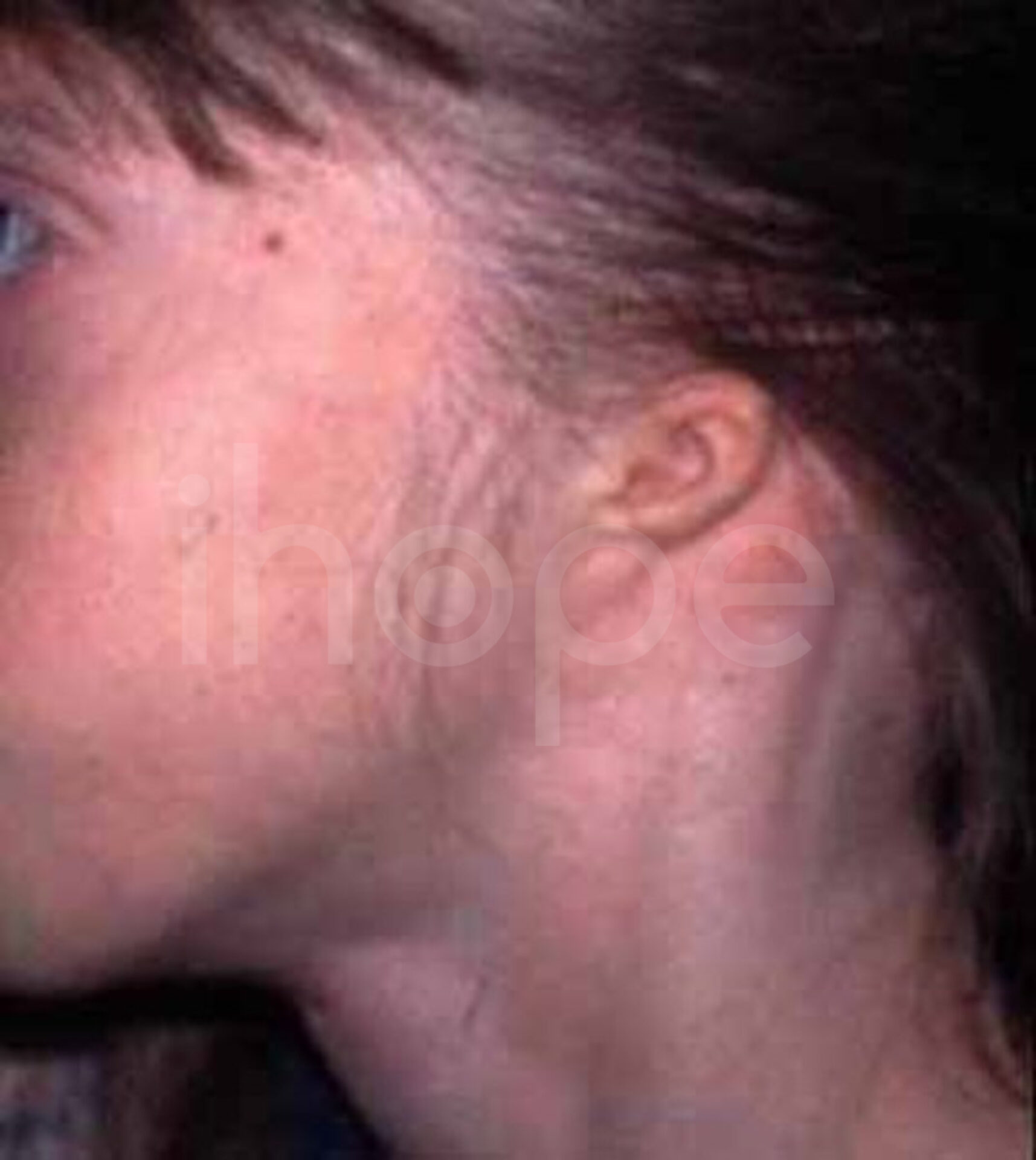
Ảnh: Tai nhỏ
Nguồn: Elements of Morphology, National Human Genome Research Institute
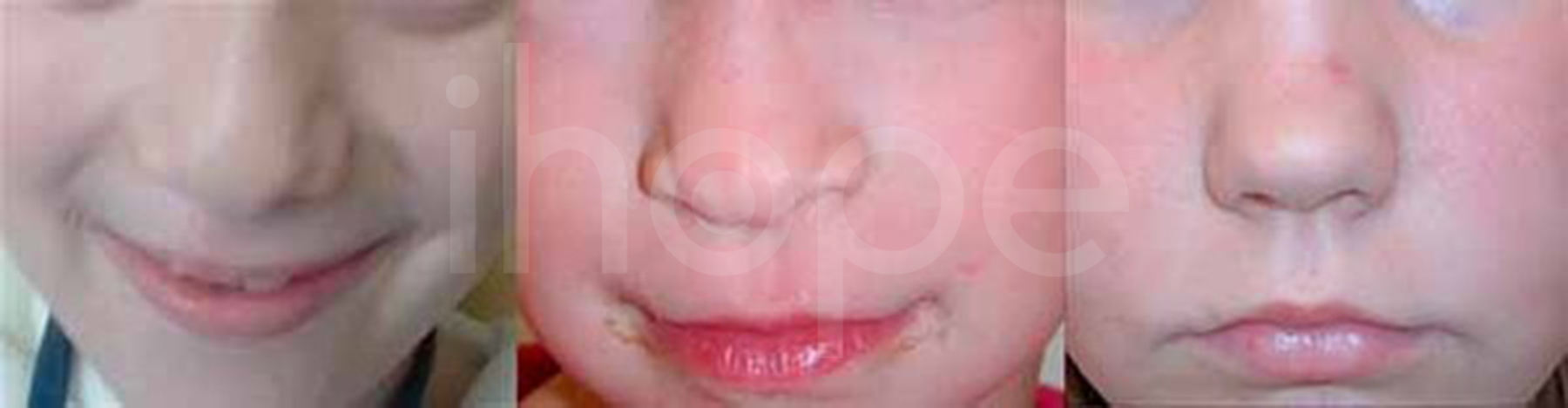
Ảnh: Mũi ngắn với đầu mũi lớn
Nguồn: Elements of Morphology, National Human Genome Research Institute
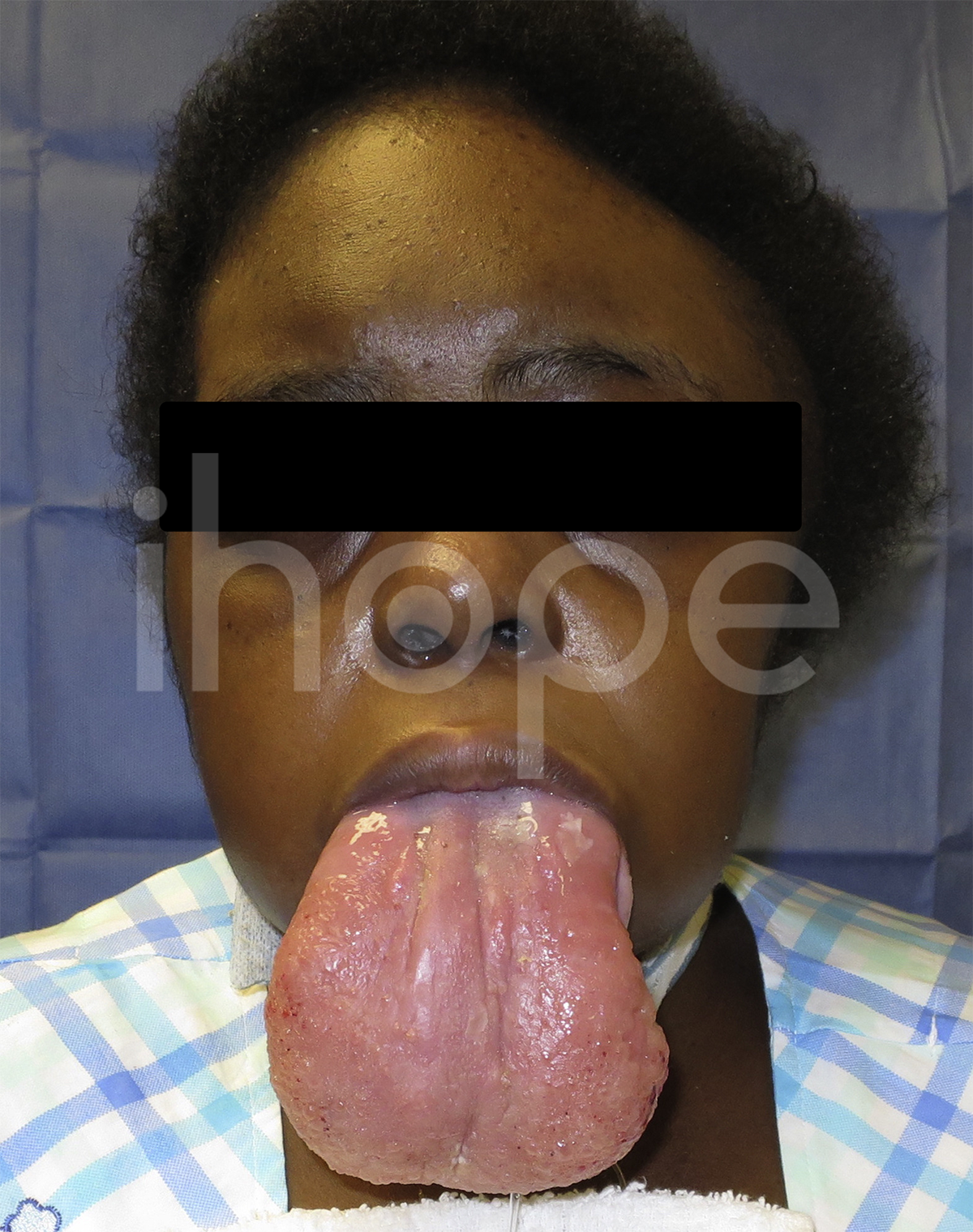
Ảnh: Lưỡi to
Nguồn: Elements of Morphology, National Human Genome Research Institute
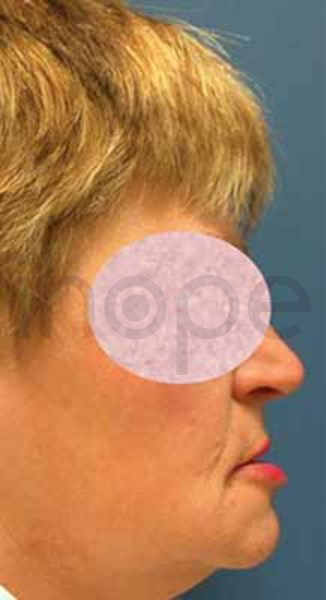
Ảnh: Cằm chìa
Nguồn: Elements of Morphology, National Human Genome Research Institute
Da, cơ, xương, khớp
Bệnh nhân có thể xuất hiện các bất thường về da như:
- Chảy xệ
- Tăng tiết bã nhờn da mặt
- Ửng đỏ
- U xơ mạch
Ngoài ra, người bệnh có biểu hiện teo cơ tiến triển hoặc các bệnh lí liên quan đến xương và khớp bao gồm:
- Chậm phát triển xương
- Cột sống, xương sườn, xương đặc và xương trụ bất thường
- Sọ dày
- Bàn chân dẹt
- Viêm bao hoạt dịch
- Viêm khớp
- Cứng khớp
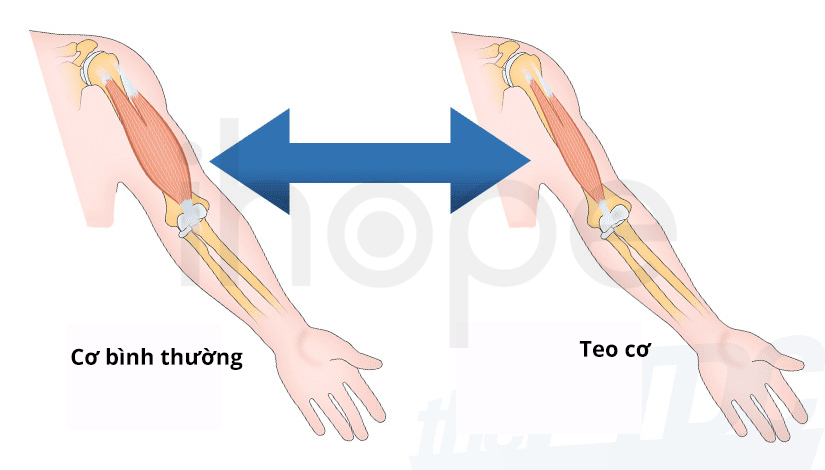
Ảnh: Teo cơ
Nguồn: Mount Sinai
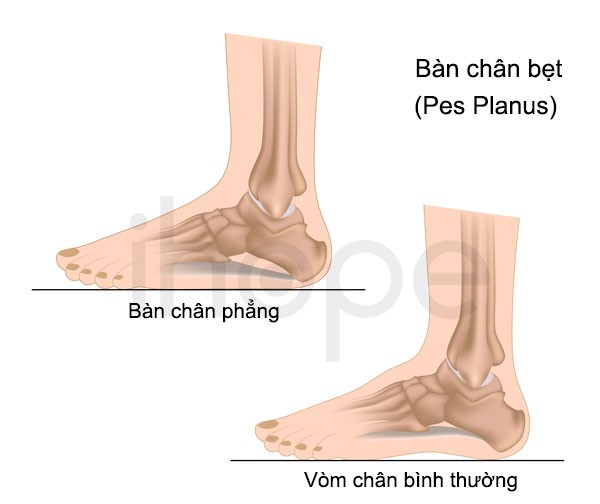
Ảnh: Bàn chân bẹt
Nguồn: U.S. National Library of Medicine
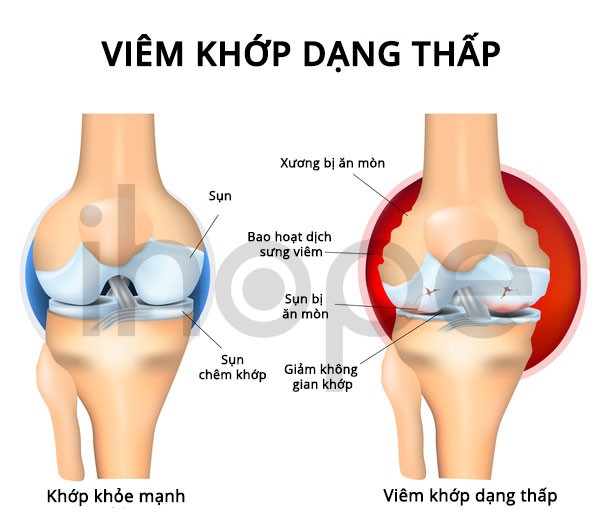
Ảnh: Viêm khớp dạng thấp
Nguồn: U.S. National Library of Medicine.
Răng miệng
Bệnh nhân thường gặp các vấn đề về răng miệng như nhiễm trùng và viêm nướu.
Một số biểu hiện khác
Một số biểu hiện khác của bệnh aspartylglucosamin niệu bao gồm:
- Thoát vị cơ quan nội tạng
- Tinh hoàn lớn
- Viêm tai giữa mãn tính
- Gan và lách to
- Kém hấp thu
- Nhiễm trùng đường hô hấp
- Rối loạn tiêu hóa
- Giảm bạch cầu trung tính và tiểu cầu
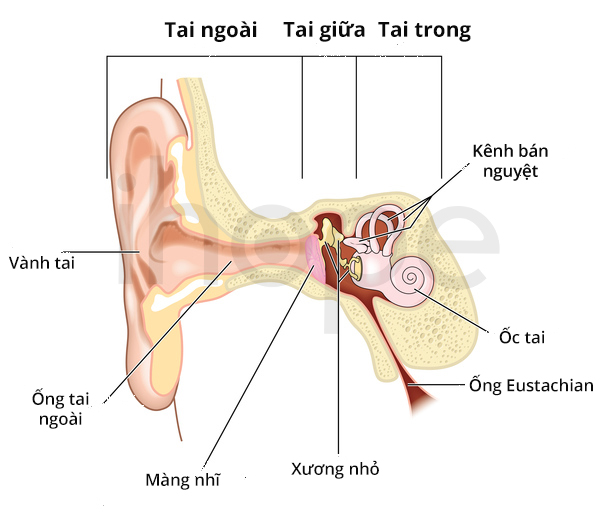
Ảnh: Cấu trúc tai bình thường
Nguồn: Blamb/Shutterstock.com
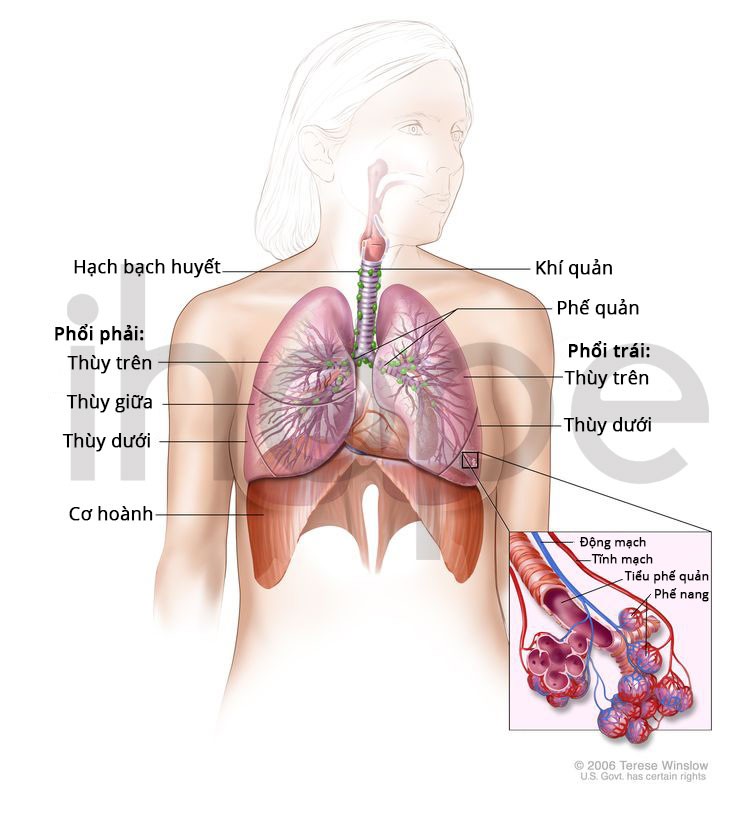
Ảnh: Cấu trúc hệ hô hấp
Nguồn: National Cancer Institute.
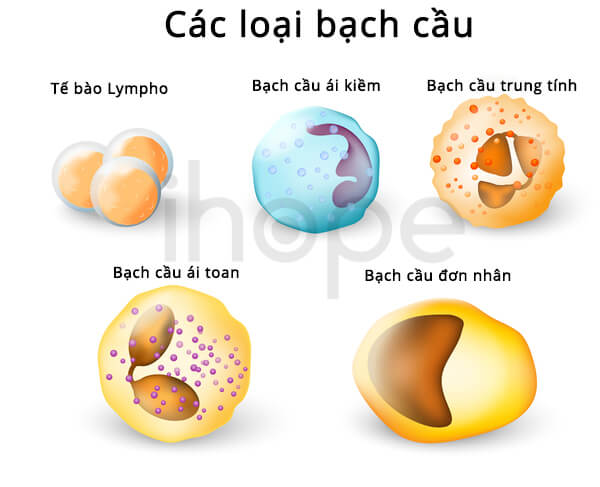
Ảnh: Các loại bạch cầu
Nguồn: Designua/Shutterstock.com
Độ phổ biến
Theo ước tính, mỗi năm tại Phần Lan có khoảng 1–3 trẻ sinh ra mắc bệnh aspartylglucosamin niệu. Bệnh ít phổ biến hơn tại những khu vực khác. Tuy nhiên, người ta chưa có thống kê cụ thể tỉ lệ mắc bệnh.
Nguyên nhân
Đột biến gen AGA gây ra bệnh aspartylglucosamin niệu. Gen này cung cấp hướng dẫn tạo ra protein aspartylglucosaminidase. Aspartylglucosaminidase là enzyme hoạt động trong lysosome—trung tâm tái chế của tế bào. Enzyme aspartylglucosaminidase có chức năng cắt liên kết giữa gốc đường N-acetylglucosamine và axit amin asparagine trong các phân tử glycoprotein.
Đột biến gen AGA gây thiếu enzyme aspartylglucosaminidase trong lysosome và ngăn cản quá trình phân cắt glycoprotein. Vì vậy, glycoprotein tích tụ trong lysosome đến mức làm gián đoạn chức năng bình thường của tế bào rồi cuối cùng dẫn đến cái chết tế bào. Nồng độ glycoprotein cao ảnh hưởng nghiêm trọng đến các tế bào thần kinh trong não, do đó người bệnh biểu hiện nhiều dấu hiệu và triệu chứng bệnh aspartylglucosamin niệu.
Chẩn đoán
Bác sĩ thường chẩn đoán bệnh aspartylglucosamin niệu thông qua xét nghiệm di truyền hoặc giải trình tự exome. Ngoài ra, bác sĩ có thể chẩn đoán bệnh bằng cách đo mức độ bài tiết aspartylglucosamin trong nước tiểu bằng phương pháp sắc ký lỏng/khối phổ hoặc đo hoạt tính của enzyme aspartylglucosaminidase trong huyết thanh, tế bào lympho, nguyên bào sợi, tế bào ối hoặc nguyên bào nuôi.
Điều trị
Hiện nay, chưa có phương pháp điều trị bệnh aspartylglucosamin niệu. Những ca ghép tủy xương đồng loại không mang lại kết quả tốt. Nếu người bệnh thực hiện liệu pháp ghép tế bào gốc tạo máu, họ cần tiến hành sớm để làm chậm quá trình suy giảm nhận thức. Ngoài ra, thuốc trimethylglycine hiện đang được đánh giá lâm sàng.
Những phương pháp sau có thể giúp cải thiện triệu chứng bệnh và giảm các biến chứng:
- Thuốc chống động kinh
- Điều trị rối loạn giấc ngủ
- Trị liệu hành vi
- Chăm sóc răng miệng hằng ngày
- Phẫu thuật cắt màng nhĩ, vòm họng hoặc amidan
- Điều trị cột sống và xương khớp
- Điều trị tiêu chảy hoặc táo bón
Dạng di truyền
Aspartylglucosamin niệu di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh lặn trên nhiễm sắc thể thường sẽ có bố và mẹ mang một bản sao của gen đột biến, nhưng họ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Bệnh aspartylglucosamin niệu di truyền lặn do đột biến gen AGA, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kỳ.
Các tên gọi khác
- AGA deficiency
- Aspartylglycosaminuria
- Glycosylasparaginase deficiency
- Aspartylglucosamidase deficiency
- Aspartylglucosaminidase deficiency
References
- Genetic Testing Information. Aspartylglucosaminuria. Retrieved May 22, 2024 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0268225/
- Genetic and Rare Diseases Information Center. Aspartylglucosaminuria. Retrieved May 22, 2024 from https://rarediseases.info.nih.gov/diseases/5854/index
- Catalog of Genes and Diseases from OMIM. ASPARTYLGLUCOSAMINURIA; AGU. Retrieved May 22, 2024 from https://omim.org/entry/208400
- U.S National Library of Medicine. Aspartylglucosaminuria. Retrieved May 22, 2024 from https://medlineplus.gov/genetics/condition/aspartylglucosaminuria/#resources
- National Institute of Health. Aspartylglucosaminuria. Retrieved May 22, 2024 from https://www.ncbi.nlm.nih.gov/sites/books/NBK599378/
- National Organization for Rare Disorders. Aspartylglycosaminuria. Retrieved May 22, 2024 from https://rarediseases.org/rare-diseases/aspartylglycosaminuria/
- Orphanet. Aspartylglucosaminuria. Retrieved May 22, 2024 from https://www.orpha.net/en/disease/detail/93?mode=name
- National Institute of Health. Aspartylglucosaminuria: Clinical Presentation and Potential Therapies. Retrieved May 22, 2024 from https://pubmed.ncbi.nlm.nih.gov/33439067/





