
Loạn sản xương hàm ảnh hưởng đến quá trình phát triển của xương, sắc tố da và quá trình phân bố chất béo. Người bệnh thường chậm phát triển sau sinh. Bệnh được chia thành hai loại dựa vào mô hình phân bố chất béo trong cơ thể.
Biểu hiện lâm sàng
Các dấu hiệu của bệnh xuất hiện ngay từ lúc trẻ mới sinh bao gồm:
- Xương hàm dưới kém phát triển khiến cằm thụt vào trong
- Xương đòn dị dạng làm vai nghiêng
- Mất xương từ các đầu ngón tay (acroosteolysis) gây ra đầu ngón tay hình củ ấu
- Các khớp xương hộp sọ đóng chậm
Loạn sản xương chia thành hai loại dựa vào triệu chứng loạn dưỡng mỡ tại một số vùng trên cơ thể, bao gồm:
- Loạn sản xương hàm phát triển loạn dưỡng mỡ loại A (mandibuloacral dysplasia with type A lipodystrophy - MADA) là loạn dưỡng mỡ một phần, người bệnh bị mất mô mỡ từ thân và tay chân. Mỡ tích tụ quanh cổ và vai. Bệnh thường khởi phát ở người trưởng thành.
- Loạn sản xương hàm phát triển loạn dưỡng mỡ loại B (mandibuloacral dysplasia with type B lipodystrophy - MADB) là loạn dưỡng mỡ toàn phần hiếm gặp, người bệnh bị mất mô mỡ ở mặt, thân và tay chân. Trẻ mắc bệnh thường sinh non và khởi phát bệnh ngay sau sinh.
Rối loạn phân bố mỡ có thể liên quan đến hội chứng chuyển hóa, bao gồm kháng insulin. Người bệnh phát triển các biến chứng như không dung nạp glucose, tăng triglycerid máu và tiểu đường.
Độ phổ biến
Chứng loạn sản xương hàm hiếm gặp, do đó hiện nay tỷ lệ mắc bệnh vẫn chưa được thống kê cụ thể.
Nguyên nhân
Hai loại loạn sản xương hàm do đột biến các gen khác nhau:
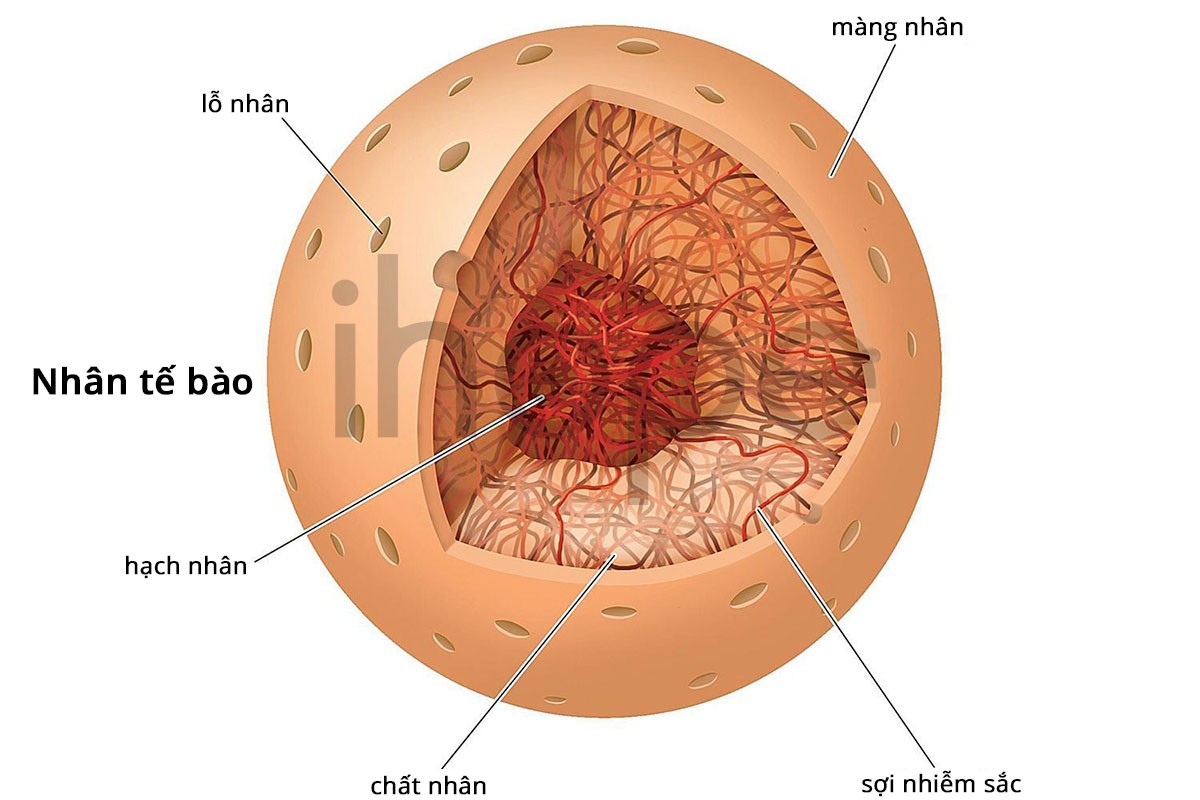
Các gen này tham gia nhiều quá trình tế bào và góp phần duy trì cấu trúc của nhân.
Nguồn: Shutterstock
Gen LMNA cung cấp hướng dẫn tạo ra protein lamin A và lamin C. Hai protein này là thành phần trong phần vỏ bao quanh nhân tế bào. Vỏ nhân kiểm soát các phân tử di chuyển ra vào nhân và điều hòa hoạt động của một số gen. Đột biến gen LMNA làm thay đổi cấu trúc của lamin A và lamin C.
Protein lamin A cần được xử lý trong tế bào trước khi trở thành một phần của vỏ nhân. Protein được tạo ra từ gen ZMPSTE24 tham gia vào quá trình xử lý này. Nó cắt protein lamin A chưa trưởng thành (prelamin A) tại một vị trí để tạo thành lamin A. Các đột biến gen ZMPSTE24 dẫn đến tích tụ prelamin A và thiếu protein trưởng thành.
Đột biến gen LMSA hoặc ZMPSTE24 có khả năng phá vỡ cấu trúc của vỏ nhân. Tuy nhiên, người ta vẫn chưa hiểu rõ cơ chế dẫn đến các triệu chứng của bệnh loạn sản xương hàm.
Chẩn đoán
Người bệnh được khám sức khỏe, thăm hỏi tiền sử bệnh gia đình và kiểm tra các dấu hiệu lâm sàng như hình thái xương hàm, tình trạng da.
Xét nghiệm di truyền tìm đột biến gen có thể chẩn đoán bệnh.
Điều trị
Hiện nay chưa có phương pháp điều trị hoàn toàn chứng loạn sản xương hàm. Các liệu pháp hỗ trợ dựa vào triệu chứng cụ thể của mỗi người bệnh và cải thiện chất lượng sống của họ.
- Người bệnh nên khám sức khỏe định kỳ nhằm tầm soát bệnh tiểu đường.
- Tập thể dục thường xuyên và duy trì cân nặng hợp lý giảm nguy cơ phát triển bệnh tiểu đường.
- Thực hiện chế độ ăn lành mạnh, hạn chế thức ăn chứa nhiều dầu mỡ.
Dạng di truyền
Bệnh di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh lặn trên nhiễm sắc thể thường sẽ có bố và mẹ mang một bản sao của gen đột biến, nhưng bố mẹ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Loạn sản xương hàm di truyền lặn trên nhiễm sắc thể thường, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con.
Các thành viên trong gia đình nên khám sức khỏe và tầm soát bệnh định kỳ nếu có thành viên mắc bệnh. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh.
Các tên gọi khác
- Mandibuloacral dysostosis
References
- Genetic Testing Information. Mandibuloacral dysplasia. Retrieved September 20, 2022 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0432291/
- Genetic Testing Information. Mandibuloacral dysplasia with type B lipodystrophy. Retrieved September 20, 2022 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C1837756/
- Genetic and Rare Diseases Information Center. Mandibuloacral dysplasia. Retrieved September 20, 2022 from https://rarediseases.info.nih.gov/diseases/11893/mandibuloacral-dysplasia/
- Catalog of Genes and Diseases from OMIM. MANDIBULOACRAL DYSPLASIA WITH TYPE A LIPODYSTROPHY. Retrieved September 20, 2022 from https://omim.org/entry/248370
- Catalog of Genes and Diseases from OMIM. MANDIBULOACRAL DYSPLASIA WITH TYPE B LIPODYSTROPHY Retrieved September 20, 2022 from https://omim.org/entry/608612
- U.S National Library of Medicine. Mandibuloacral dysplasia. Retrieved September 20, 2022 from https://medlineplus.gov/genetics/condition/mandibuloacral-dysplasia/
- National Organization for Rare Disorders. Mandibuloacral Dysplasia. Retrieved September 20, 2022 from https://rarediseases.org/rare-diseases/mandibuloacral-dysplasia/
- Orphanet. Mandibuloacral Dysplasia. Retrieved September 20, 2022 from https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=2457
- Vittoria Cenniab, Maria Rosaria D’Apicec, Paolo Garagnanide, Marta Columbarob, Giuseppe Novellicf, Claudio Franceschid, Giovanna Lattanziab. Mandibuloacral dysplasia: A premature ageing disease with aspects of physiological ageing. Published March, 2018 https://doi.org/10.1016/j.arr.2017.12.001
- M. M. Hitzert, S. N. van der Crabben, G. Baldewsingh, H. K. Ploos van Amstel, A. van den Wijngaard, C. M. A. van Ravenswaaij-Arts & C. W. R. Zijlmans. Mandibuloacral dysplasia type B (MADB): a cohort of eight patients from Suriname with a homozygous founder mutation in ZMPSTE24 (FACE1), clinical diagnostic criteria and management guidelines. Published December 19, 2019 https://ojrd.biomedcentral.com/articles/10.1186/s13023-019-1269-0





