
Dậy thì sớm ở nam giới mang tính gia đình (familial male-limited precocious puberty) là bệnh di truyền. Nam giới mắc bệnh dậy thì sớm, khoảng từ 2 đến 5 tuổi. Bệnh không ảnh hưởng đến nữ giới.
Biểu hiện lâm sàng
Trẻ mắc bệnh xuất hiện các dấu hiệu dậy thì, bao gồm:
- Giọng trầm
- Mụn trứng cá
- Mọc nhiều lông
- Có mùi hôi nách
- Dương vật và tinh hoàn phát triển sớm
- Cương cứng tự phát
Trẻ bắt đầu thay đổi tâm lý và hành vi như hung hăng và quan tâm đến tình dục.
Nếu không điều trị, trẻ sẽ phát triển nhanh rồi ngừng sớm hơn bình thường. Do đó, người bệnh khi trưởng thành có xu hướng thấp hơn các thành viên khác trong gia đình.
Độ phổ biến
Dậy thì sớm ở nam giới mang tính gia đình hiếm gặp, do đó hiện nay tỷ lệ mắc bệnh vẫn chưa được thống kê cụ thể.
Nguyên nhân
Đột biến gen LHCGR gây ra dậy thì sớm ở nam giới mang tính gia đình.
Gen LHCGR cung cấp hướng dẫn tạo ra protein thụ thể gonadotropin màng đệm (hormone tạo hoàng thể). Trên protein thụ thể, có một số vị trí cho protein ligand (phối tử) gắn vào tương tự như chìa khoá và ổ khoá. Chúng liên kết với nhau nhằm kích hoạt tín hiệu phát triển và thực hiện chức năng của tế bào.
Protein tạo ra từ gen LHCGR hoạt động như thụ thể cho hai phối tử hormone tạo hoàng thể (luteinizing) và hormone gonadotropin màng đệm. Thụ thể giúp cơ thể đáp ứng với hai loại hormone này. Đối với nam giới, gonadotropin màng đệm kích thích quá trình phát triển tế bào Leydig trong tinh hoàn và hormone luteinizing kích hoạt tế bào Leydig sản xuất nội tiết tố nam. Androgen và testosterone là hormone kiểm soát quá trình phát triển và sinh sản của nam giới. Đối với nữ, hormone luteinizing kích thích rụng trứng, gonadotropin màng đệm được sản xuất trong giai đoạn mang thai giúp duy trì các điều kiện cần thiết cho thai kỳ.
Đột biến gen LHCGR khiến protein thụ thể kích hoạt liên tục dù không liên kết với hormone tạo hoàng thể hoặc gonadotropin màng đệm. Thụ thể hoạt động quá mức gây tăng sản testosterone dẫn đến dậy thì sớm ở nam giới. Các thụ thể hoạt động quá mức không ảnh hưởng đến phụ nữ.
Khoảng 18% người bệnh không mang đột biến gen LHCGR. Nguyên nhân mắc bệnh của những trường hợp này chưa rõ ràng.
Chẩn đoán
Bệnh được chẩn đoán bệnh dựa trên biểu hiện lâm sàng, tiền sử bệnh của cá nhân và gia đình. Nam giới nghi ngờ mắc bệnh cần thực hiện các xét nghiêm bổ sung nhằm hỗ trợ chẩn đoán và phân biệt với các bệnh lý khác.
Các xét nghiệm có thể thực hiện bao gồm:
- Chụp X-quang đánh giá mức độ phát triển của xương
- Xét nghiệm sinh hóa đánh giá nồng độ testosterone huyết thanh, gonadotropin
- Xét nghiệm di truyền tìm đột biến gen LHCGR gây bệnh
Điều trị
Mục tiêu điều trị ngăn chặn tăng trưởng thể chất quá nhanh, trì hoãn thời điểm bắt đầu hành kinh, đưa chiều cao cuối cùng của người bệnh nhân gần hơn với kỳ vọng di truyền.
Hai phương pháp được đề xuất bao gồm:
- Sử dụng bicalutamide đối kháng androgen cùng các chất ức chế aromatase (anastrozole hoặc letrozole) để bình thường hóa tốc độ tăng trưởng cho đến khi đạt được chiều cao trưởng thành
- Sử dụng các chất ức chế sinh tổng hợp androgen (ketoconazole) làm giảm nồng độ testosterone
Trường hợp dậy thì sớm trung ương (phụ thuộc vào gonadotropin), bác sĩ có thể bổ sung thêm liệu pháp GnRH.
Dạng di truyền
Bệnh di truyền theo kiểu trội trên nhiễm sắc thể thường và chỉ xảy ra ở nam giới. Do đó, một bản sao của gen LHCGR trong mỗi tế bào đủ để gây ra bệnh. Người bệnh có thể thừa hưởng gen đột biến từ cha hoặc mẹ mắc bệnh hoặc do đột biến gen LHCGR mới ở gia đình không có tiền sử bệnh trong gia đình của họ.

Ảnh: Sơ đồ di truyền gen trội từ cha mẹ sang con
Nguồn: U.S. National Library of Medicine
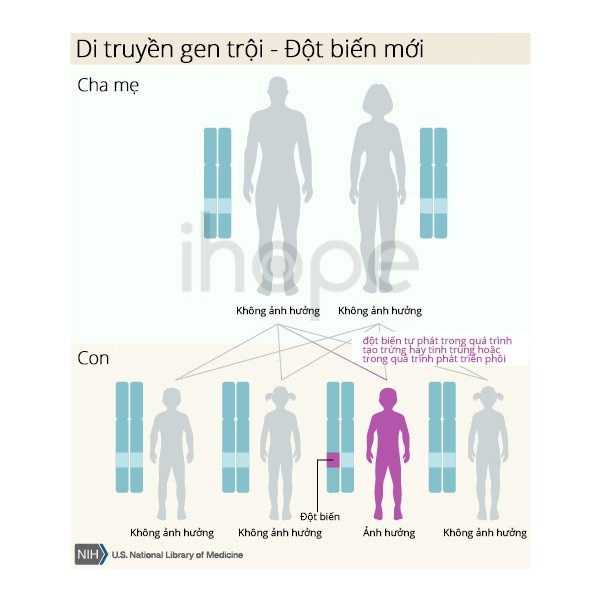
Ảnh: /wp-content/uploads/2020/09/di-truyen-gen-troi.jpeg" alt="Di truyền gen trội
Nữ giới mang đột biến gen gây bệnh không biểu hiện các triệu chứng của bệnh.
Phòng ngừa
Nếu nam giới mắc bệnh, khi sinh con sẽ có 50% khả năng di truyền bệnh. Do đó, để bảo đảm 100% khả năng con không bị bệnh, cha mẹ có thể chọn phương pháp thụ tinh nhân tạo IVF và sàng lọc phôi PGS/PGD. Các cặp vợ chồng trước khi mang thai cần tư vấn di truyền đảm bảo sinh con khỏe mạnh.
Các tên gọi khác
- Familial gonadotrophin-independent sexual precocity GIPP
- Gonadotrophin-independent precocious puberty
- Precocious pseudopuberty
- Pubertas praecox
- Testotoxicosis
References
- Genetic Testing Information. Gonadotropin-independent familial sexual precocity. Retrieved February 20, 2023 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0342549/
- Genetic and Rare Diseases Information Center. Precocious puberty. Retrieved February 20, 2023 from https://rarediseases.info.nih.gov/diseases/7446/precocious-puberty
- Catalog of Genes and Diseases from OMIM. PRECOCIOUS PUBERTY, MALE-LIMITED. Retrieved February 20, 2023 from https://omim.org/entry/176410
- U.S National Library of Medicine. Familial male-limited precocious puberty. Retrieved February 20, 2023 from https://medlineplus.gov/genetics/condition/familial-male-limited-precocious-puberty/
- MalaCards. Precocious Puberty, Male-Limited (FMPP). Retrieved February 20, 2023 from https://www.malacards.org/card/precocious_puberty_male_limited
- National Institute of Health. A Case of Familial Male-limited Precocious Puberty with a Novel Mutation. Retrieved February 20, 2023 from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8186329/
- National Organization for Rare Disorders. Precocious Puberty. Retrieved February 20, 2023 from https://rarediseases.org/rare-diseases/precocious-puberty/
- Orphanet. Familial male limited precocious puberty. Retrieved February 20, 2023 from https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=3000
- Brito VN, Latronico AC, Arnhold IJ, Mendonca BB. Update on the etiology, diagnosis and therapeutic management of sexual precocity. Arq Bras Endocrinol Metabol. 2008 Feb;52(1):18-31. doi: 10.1590/s0004-27302008000100005
- Soriano-Guillen L, Mitchell V, Carel JC, Barbet P, Roger M, Lahlou N. Activating mutations in the luteinizing hormone receptor gene: a human model of non-follicle-stimulating hormone-dependent inhibin production and germ cell maturation. J Clin Endocrinol Metab. 2006 Aug;91(8):3041-7. doi: 10.1210/jc.2005-2564





