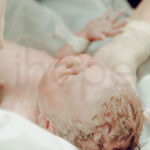
Hội chứng Carpenter (Carpenter syndrome) là hội chứng di truyền hiếm gặp với một số biểu hiện đặc trưng gồm xương sọ dung hợp sớm (craniosynostosis), bất thường ngón tay và ngón chân cùng với các vấn đề phát triển khác.
Biểu hiện lâm sàng
Sự dung hợp xương sọ sớm cản trở hộp sọ phát triển bình thường, khiến đầu có hình dáng nhọn (acrocephaly). Trên những người bệnh có biểu hiện nặng, hộp sọ biến dạng thành hình cỏ ba lá. Quá trình dung hợp này gây ra mất cân xứng sọ mặt, ảnh hưởng đến phát triển não bộ và làm tăng áp lực nội sọ.
Bệnh nhân mắc hội chứng Carpenter thường mang các đặc điểm dị biệt khuôn mặt bao gồm:
- Sống mũi tẹt
- Mắt cụp xuống
- Tai thấp và có hình dáng bất thường
- Hàm trên và hàm dưới kém phát triển
- Bất thường về hình dáng mắt và suy giảm thị lực
- Bất thường về răng, bao gồm răng sữa kích thước nhỏ
Một số bệnh nhân có các bất thường ngón tay và ngón chân bao gồm:
- Dính ngón, phổ biến nhất giữa ngón tay thứ ba và thứ tư
- Ngón tay hoặc ngón chân ngắn bất thường
- Thừa ngón thường xuất hiện gần ngón chân cái, ngón chân thứ hai hoặc ngón tay thứ năm
Thiểu năng trí tuệ khá phổ biến trên những người mắc hội chứng, dao động từ mức độ nhẹ đến nặng. Tuy nhiên, vẫn có một số trường hợp mắc bệnh có trí tuệ bình thường. Nguyên nhân gây triệu chứng này chưa được xác định, do mức độ nghiêm trọng của dị tật sọ não không liên quan đến mức độ suy giảm trí tuệ.
Các triệu chứng khác bao gồm:
- Béo phì khởi phát từ giai đoạn trẻ nhỏ
- Thoát vị rốn
- Mất thính lực và dị tật tim
- Dị tật xương như biến dạng khớp háng, gù vẹo cột sống và đầu gối vẹo trong
- Phần lớn nam giới mắc bệnh có bất thường sinh dục, phổ biến nhất là tinh hoàn ẩn
Một số bệnh nhân có nội tạng bất thường về vị trí và cấu trúc bao gồm các cơ quan nội tạng bị đảo ngược hoàn toàn so với vị trí bình thường (situs inversus), tim nằm lệch sang bên phải thay vì bên trái (dextrocardia) hoặc rối loạn lưu thông máu do các bất thường trên hệ thống động mạch lớn.
Tuổi thọ của người bệnh bị rút ngắn nhưng thay đổi tùy theo từng cá nhân. Hội chứng Carpenter có nhiều triệu chứng tương đồng với hội chứng Greig cephalopolysyndactyly, bao gồm dính khớp sọ, thừa ngón và bất thường tim mạch, do đó bệnh nhân cần xét nghiệm di truyền để chẩn đoán chính xác.
Độ phổ biến
Hội chứng Carpenter là một bệnh hiếm gặp. Khoảng 70 trường hợp mắc bệnh đã được báo cáo trong các tài liệu khoa học.
Nguyên nhân
Hội chứng Carpenter bắt nguồn từ đột biến gen RAB23 hoặc MEGF8. Gen RAB23 cung cấp hướng dẫn sản xuất ra protein có nhiều chức năng gồm tham gia quá trình vận chuyển túi ngoại bào, điều hướng các protein và phân tử bên trong tế bào. Protein Rab23 vận chuyển túi màng từ màng tế bào đến vị trí thích hợp nhằm vận chuyển vật liệu cần thiết kích hoạt truyền tín hiệu trong quá trình phát triển. Chẳng hạn, protein Rab23 điều chỉnh đường tín hiệu hedgehog—tín hiệu thiết yếu đối với quá trình tăng sinh, biệt hóa tế bào và định hình bình thường của nhiều bộ phận cơ thể.
Gen MEGF8 cung cấp hướng dẫn tạo ra protein có chức năng chưa rõ ràng. Dựa trên cấu trúc, protein Megf8 có khả năng tham gia vào quá trình kết dính tế bào và hỗ trợ tương tác giữa các protein. Các nhà nghiên cứu cũng nghi ngờ protein Megf8 nắm vai trò định hình cơ thể bình thường.
Đột biến gen RAB23 hoặc MEGF8 dẫn đến các phiên bản protein mất chức năng hoặc giảm chức năng nghiêm trọng. Mặc dù cơ chế chính xác chưa rõ ràng, sự can thiệp vào quá trình định hình cơ thể giữ vai trò cốt lõi. Người mang đột biến gen MEGF8 có nguy cơ cao mắc bệnh đảo ngược vị trí tim và các bất thường vị trí cơ quan khác, tuy nhiên họ có mức độ dính khớp sọ nhẹ hơn so với những người mang đột biến gen RAB23.
Chẩn đoán
Quá trình chẩn đoán hội chứng Carpenter đôi khi được đề xuất trước sinh (tiền sản) dựa trên các xét nghiệm chuyên sâu như nội soi thai nhi hoặc siêu âm. Thông qua nội soi thai nhi, bác sĩ đưa ống nội soi vào tử cung qua thành bụng nhằm quan sát trực tiếp, đồng thời lấy mẫu máu hoặc mô thai nhi. Siêu âm thai nhi là phương pháp chẩn đoán không xâm lấn bằng sóng âm phản xạ để tạo ra hình ảnh thai nhi. Xét nghiệm di truyền trước sinh hỗ trợ xác nhận chẩn đoán thông qua mẫu mô thai nhi được lấy từ sinh thiết gai nhau hoặc chọc ối.
Đa số các trường hợp được chẩn đoán hoặc xác nhận ngay khi sinh hoặc ngay sau sinh thông qua khám lâm sàng toàn diện, xác định các dấu hiệu thể chất điển hình và nhiều xét nghiệm chuyên sâu khác. Các xét nghiệm này bao gồm chụp cắt lớp vi tính (CT) hoặc chụp cộng hưởng từ (MRI) nhằm phát hiện dị tật xương và thính giác. Chụp CT kết hợp tia X tạo hình ảnh cắt ngang các cấu trúc bên trong. MRI sử dụng từ trường và sóng vô tuyến tạo hình ảnh mặt cắt ngang chi tiết của các cơ quan và mô.
Xét nghiệm đột biến gen RAB23 và MEGF8 có thể xác nhận chẩn đoán lâm sàng. Ngoài ra, kiểm tra tim mạch toàn diện được khuyến nghị nhằm phát hiện các bất thường, bao gồm khám lâm sàng, nghe tim phổi, chụp X-quang, điện tâm đồ (EKG), siêu âm tim và thông tim.
Điều trị
Hiện nay, chưa có phương pháp điều trị đặc hiệu cho hội chứng Carpenter, bệnh nhân chủ yếu được chữa theo triệu chứng cụ thể. Can thiệp phẫu thuật sớm có thể ngăn ngừa hoặc điều chỉnh tình trạng khớp sọ đóng sớm, đồng thời làm giảm áp lực nội sọ. Phẫu thuật sớm hỗ trợ ngăn ngừa thiểu năng trí tuệ trong một vài trường hợp. Tuy nhiên, không phải mọi trường hợp đều có kết quả như kì vọng. Một số bệnh nhân dù đã được phẫu thuật vẫn bị thiểu năng trí tuệ, trong khi một số người chưa từng phẫu thuật lại có trí thông minh hoàn toàn bình thường.
Phẫu thuật chỉnh hình và tái tạo được đề xuất nhằm điều chỉnh các dị tật sọ mặt, thừa ngón, dính ngón và các khiếm khuyết xương khác. Đối với bệnh nhân mắc dị tật tim bẩm sinh, bác sĩ chỉ định dùng thuốc hoặc phẫu thuật tùy thuộc vào mức độ nghiêm trọng và vị trí các bất thường giải phẫu.
Máy trợ thính và liệu pháp ngôn ngữ mang lại lợi ích cho người bệnh suy giảm thính lực và gặp vấn đề phát âm. Can thiệp sớm rất quan trọng để trẻ em mắc hội chứng Carpenter có thể phát triển tối đa tiềm năng.
Tư vấn di truyền được khuyến nghị cho bệnh nhân và gia đình. Đánh giá chẩn đoán dành cho các thành viên trong gia đình giữ vai trò quan trọng nhằm phát hiện triệu chứng và các đặc điểm thể chất liên quan.
Dạng di truyền
Hội chứng Carpenter di truyền theo kiểu lặn trên nhiễm sắc thể thường nên cả hai bản sao của gen trong mỗi tế bào đều mang đột biến. Cha mẹ của một cá nhân mắc bệnh lặn trên nhiễm sắc thể thường mỗi người mang một bản sao của gen bị đột biến, nhưng họ thường không có dấu hiệu và triệu chứng của bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Hội chứng Carpenter di truyền lặn, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn cho tương lai của con. Các thành viên trong gia đình nên khám sức khỏe và tầm soát bệnh định kì nếu có thành viên mắc bệnh. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh.
Các tên gọi khác
- ACPS II
- Acrocephalopolysyndactyly 2
- Acrocephalopolysyndactyly type II
- Acrocephalosyndactyly, type II
- Type II acrocephalosyndactyly
References
- National Library of Medicine. Carpenter syndrome. Retrieved June 16, 2026 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C1275078/
- National Organization for Rare Disorders (2026). Carpenter Syndrome. Retrieved June 16, 2026 from https://rarediseases.org/rare-diseases/carpenter-syndrome/
- OMIM. CARPENTER SYNDROME 1; CRPT1. Retrieved June 16, 2026 from https://omim.org/entry/201000
- MedlinePlus. Carpenter syndrome. Retrieved June 16, 2026 from https://medlineplus.gov/genetics/condition/carpenter-syndrome/