
Tăng glycine máu không nhiễm ceton (nonketotic hyperglycinemia) là bệnh di truyền khiến cơ thể người bệnh tăng nồng độ glycine. Glycine là một axit amin cấu tạo nên protein và dẫn truyền xung thần kinh. Lượng glycine dư thừa tích tụ trong các mô và cơ quan dần đến mức gây bệnh, đặc biệt các vấn đề nghiêm trọng về thần kinh.
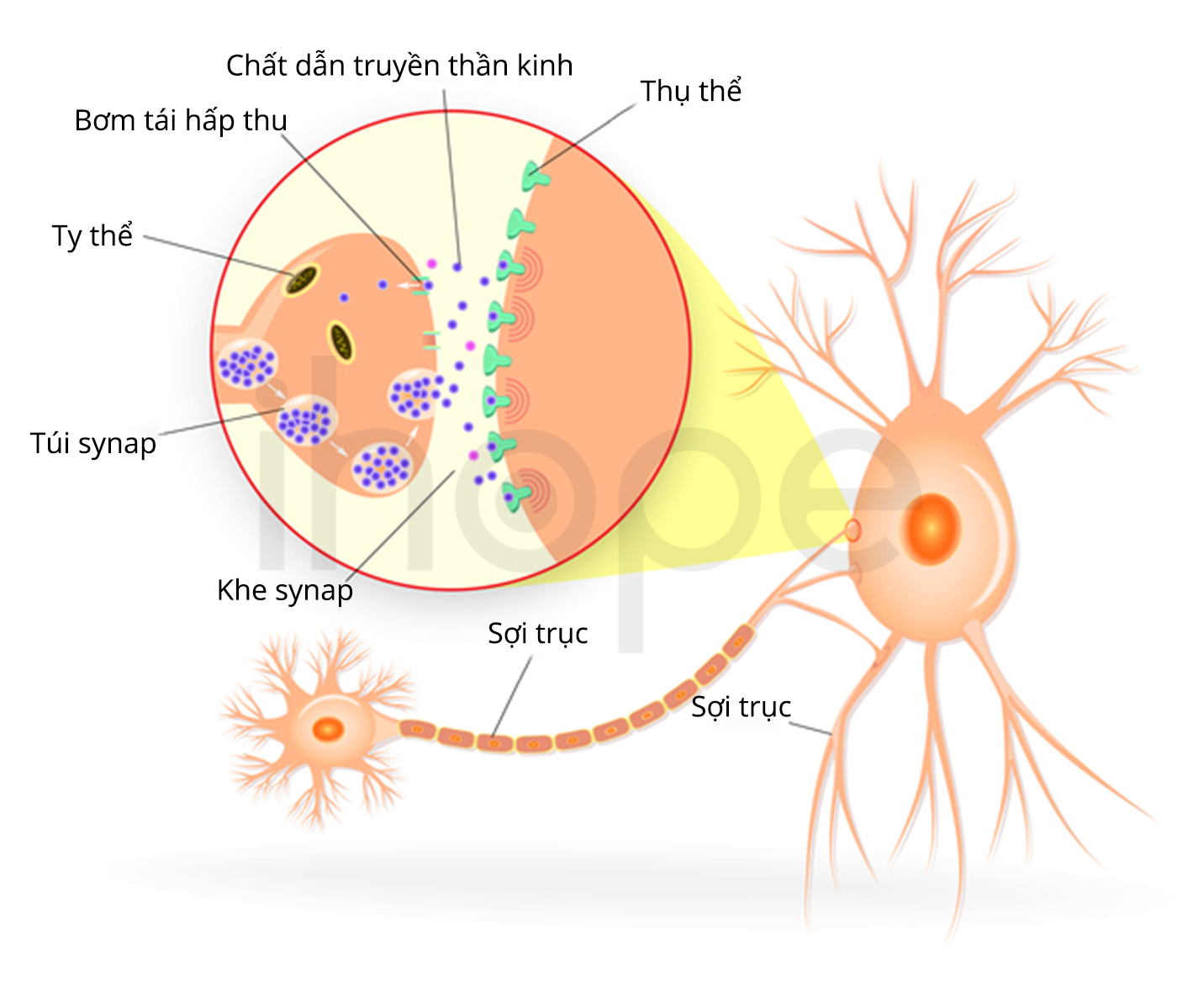
Nguồn: Designua/Shutterstock.com
Biểu hiện lâm sàng
Dựa vào mức độ nghiêm trọng của bệnh, tăng glycine máu không nhiễm ceton được chia thành 2 dạng gồm thể nặng và thể nhẹ. Cả hai dạng này đều khởi phát sau sinh, phần lớn ở thể nặng. Trẻ mắc bệnh thường buồn ngủ cực độ dẫn đến hôn mê sâu. Trẻ có thể bị yếu cơ rõ rệt kèm theo các vấn đề hô hấp đe dọa tính mạng trong tuần đầu sau sinh.
Nếu sống sót sau những dấu hiệu và triệu chứng ban đầu, trẻ phát triển chứng co cứng cơ, khó bú, thiểu năng trí tuệ và co giật mất kiểm soát. Trẻ khó đạt được các mốc phát triển bình thường như bú bình, ngồi hoặc cầm nắm đồ vật. Trẻ mắc bệnh thể nhẹ phát triển chậm nhưng có thể học cách đi lại, cầm nắm, tương tác với người khác và dùng ngôn ngữ ký hiệu. Các cơn co giật nhẹ có thể được điều trị. Trẻ có thể thêm một số biểu hiện bệnh khác như cử động giật không chủ ý và hiếu động thái quá.
Độ phổ biến
Người ta ước tính tỉ lệ mắc chứng tăng glycine máu không nhiễm ceton vào khoảng 1/76.000 người trên toàn thế giới. Tỉ lệ này phổ biến hơn tại Phần Lan khoảng 1/55.000 và ở Anh, Columpia, Canada vào khoảng 1/63.000.
Nguyên nhân
Đột biến gen GLDC hoặc AMT gây tăng glycine máu không nhiễm ceton. Khoảng 80% số người bệnh bị đột biến gen GLDC. Gen GLDC và AMT cung cấp hướng dẫn tạo ra nhóm các enzyme hoạt động cùng nhau được gọi là hệ thống phân cắt glycine. Glycine là axit amin tham gia cấu tạo nên protein, chúng cũng truyền tín hiệu hóa học trong não. Khi không còn hoạt động, glycine được hệ thống enzyme phân cắt thành nhiều mảnh nhỏ hơn. Ngoài ra, quá trình này còn tạo ra các nhóm methyl để gắn vào folate. Folate là vitamin cần thiết cho tế bào và các giai đoạn phát triển não.
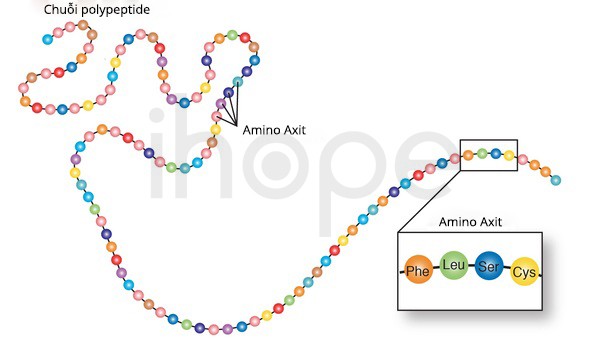
Amino axit là tập hợp 20 phân tử khác nhau, cấu tạo thành protein. Protein bao gồm một hoặc nhiều chuỗi amino axit gọi là polypeptit. Các chuỗi này xoắn cuộn thành các protein có hoạt tính sinh học khác nhau. Trình tự amino-axit được mã hóa trong gen.
Nguồn: U.S National Library of Medicine
Đột biến gen GLDC và AMT khiến hệ thống phân cắt enzyme giảm hoặc mất hoàn toàn chức năng, từ đó glycine tích tụ nhiều và folate thiếu hụt, dẫn đến các dấu hiệu và triệu chứng bệnh. Khả năng hoạt động của hệ thống phân cắt glycine quy định mức độ nghiêm trọng của bệnh.
Người mắc bệnh nhưng không mang đột biến gen GLDC hoặc AMT có thể liên quan đến các gen gồm LIAS, BOLA3, GLRX5, NFU1, ISCA2, IBA56, LIPT1 và LIPT2.
Chẩn đoán
Người bệnh được khám sức khỏe, thăm hỏi tiền sử bệnh gia đình và kiểm tra một số triệu chứng lâm sàng khởi phát sớm như yếu cơ, co giật, khó thở, hôn mê. Tuy nhiên các triệu chứng trên cũng phổ biến ở một số bệnh tương tự.
Các xét nghiệm hỗ trợ chẩn đoán bao gồm:
- Xét nghiệm máu (kiểm tra nồng độ glycine)
- Xét nghiệm nước tiểu
- Xét nghiệm dịch não tủy
- Xét nghiệm hình ảnh với trẻ mắc thể nặng, hình ảnh chụp cộng hưởng từ (MRI) cho thấy mô liên kết bán cầu não trái và phải (thể chai) nhỏ hơn mức trung bình
- Xét nghiệm di truyền tìm đột biến gen gây bệnh
Điều trị
Hiện nay chưa có phương pháp điều trị hoàn toàn bệnh tăng glycine máu không nhiễm ceton. Các liệu pháp hỗ trợ dựa vào triệu chứng cụ thể của mỗi người bệnh nhằmà cải thiện chất lượng cuộc sống của họ.
- Natri benzoat giảm nồng độ glycine trong huyết thanh, điều trị chứng co giật và giúp trẻ tỉnh táo. Benzoat liên kết với glycine trong cơ thể tạo thành hippurat rồi bài tiết qua nước tiểu. Nồng độ glycine trong huyết tương cần được theo dõi chặt chẽ nhằm đảm bảo lượng natri benzoat ở mức vừa đủ và không gây độc.
- Dextromethorphan giảm co giật và cải thiện chứng buồn ngủ. Ở người bệnh, các thụ thể NMDA trong não bị kích thích quá mức. Glutamate là chất dẫn truyền thần kinh chủ yếu liên kết với các thụ thể này. Dextromethorphan liên kết với thụ thể NMDA và ngăn không cho glutamate liên kết với thụ thể. Ketamine là chất ức chế thụ thể NMDA, nó có tác dụng tương tự dextromethorpha. Trẻ mắc bệnh thể nhẹ sử dụng dextromethorphan kết hợp với benzoat có thể cải thiện quá trình phát triển và chứng co giật.
Dạng di truyền
Chứng tăng glycine máu không nhiễm ceton di truyền lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh lặn trên nhiễm sắc thể thường sẽ có bố và mẹ mang một bản sao của gen đột biến, nhưng bố mẹ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Một số trường hợp, bệnh do đột biến mới xuất hiện trong quá trình phát sinh giao tử (trứng hoặc tinh trùng) gọi là đột biến denovo và xảy ra ở những người không có tiền sử mắc chứng rối loạn này trong gia đình họ.
Phòng ngừa
Bệnh tăng glycine máu không nhiễm ceton di truyền lặn trên nhiễm sắc thể thường, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con.
Các thành viên trong gia đình nên khám sức khỏe và tầm soát bệnh định kỳ nếu có thành viên mắc bệnh. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh.
Các tên gọi khác
- Glycine encephalopathy
- NKH
- Non-ketotic hyperglycinemia
References
- Genetic Testing Information. Non-ketotic hyperglycinemia. Retrieved September 23, 2022 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0751748/
- Genetic and Rare Diseases Information Center. Glycine encephalopathy. Retrieved September 23, 2022 from https://rarediseases.info.nih.gov/diseases/7219/glycine-encephalopathy
- Catalog of Genes and Diseases from OMIM. GLYCINE ENCEPHALOPATHY. Retrieved September 23, 2022 from https://omim.org/entry/605899
- U.S National Library of Medicine. Non-ketotic hyperglycinemia. Retrieved September 23, 2022 from https://medlineplus.gov/genetics/condition/nonketotic-hyperglycinemia/
- National Organization for Rare Disorders. Nonketotic Hyperglycinemia. Retrieved September 23, 2022 from https://rarediseases.org/rare-diseases/nonketotic-hyperglycinemia/
- Johan LK Van Hove, MD, PhD, Curtis Coughlin, II, MS, MBe, Michael Swanson, PhD, and Julia B Hennermann, MD, PhD. Nonketotic Hyperglycinemia. Published May 23, 2019 https://www.ncbi.nlm.nih.gov/books/NBK1357/
- Verywellhealth. Nonketotic Hyperglycinemia. Retrieved September 23, 2022 from https://www.verywellhealth.com/nonketotic-hyperglycinemia-overview-4176827
- Derek A.Applegarth, Jennifer R.Toone. Nonketotic Hyperglycinemia (Glycine Encephalopathy): Laboratory Diagnosis. Published September, 2021 https://doi.org/10.1006/mgme.2001.3224
- Argirios Dinopoulos, Yoichi Matsubara, Shigeo Kure. Atypical variants of nonketotic hyperglycinemia. Published September - October, 2021 https://doi.org/10.1016/j.ymgme.2005.07.016





