
Bệnh cơ mũ (cap myopathy) thuộc nhóm bệnh lí bẩm sinh về cơ hiếm gặp làm cơ người bệnh có hình dạng giống chiếc mũ. Do đó, người bệnh có biểu hiện yếu cơ và có khuôn mặt dị biệt.
Nguồn: Medlineplus.gov
Biểu hiện lâm sàng
Bệnh cơ mũ chủ yếu ảnh hưởng đến cơ xương—cấu trúc giúp cơ thể di chuyển. Bệnh nhân có dấu hiệu yếu cơ, giảm trương lực cơ toàn thân, chủ yếu tại vị trí mặt, cổ, tay và chân. Các triệu chứng thường khởi phát khi mới sinh hoặc trong giai đoạn trẻ nhỏ, chúng tiến triển chậm nhưng có thể gây nguy hiểm đến tính mạng.

Nguồn: Icahn School of Medicine at Mount Sinai
Những triệu chứng khác bao gồm:
- Trẻ sơ sinh khó nuốt
- Chậm phát triển các kĩ năng vận động như ngồi, bò, đứng, đi
- Mệt mỏi
- Dễ ngã
- Chạy, nhảy, leo cầu thang khó khăn
- Khó thở
Một số bệnh nhân có những điểm dị biệt trên khuôn mặt:
- Vòm miệng cao
- Mí mắt sụp
- Mặt dài bất thường
Ngoài ra, một số người bệnh có biểu hiện võng lưng hoặc cong vẹo cột sống . Mức độ nghiêm trọng của bệnh dựa trên số lượng cơ có cấu trúc dạng mũ. Trường hợp 70–75% tế bào cơ có cấu trúc mũ, người bệnh thường mắc các bệnh lí hô hấp nghiêm trọng và tử vong trong giai đoạn trẻ nhỏ. Nếu cấu trúc mũ hiện diện trong 10–30% tế bào cơ, các triệu chứng sẽ nhẹ hơn và người bệnh có thể sống đến giai đoạn trưởng thành.
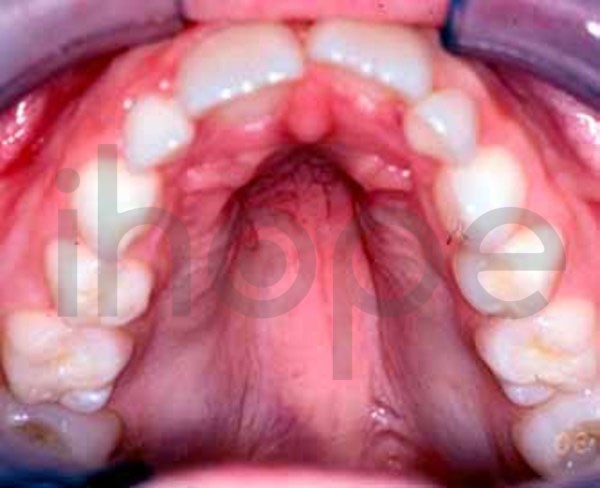
Ảnh: Vòm miệng cao
Nguồn: U.S. National Library of Medicine
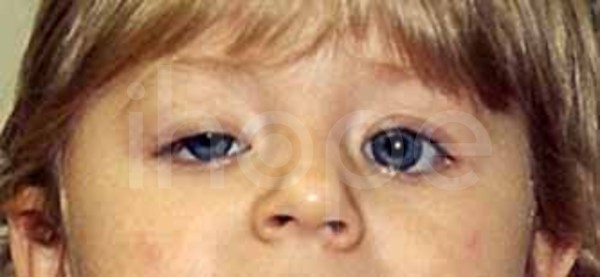
Ảnh: Mí mắt sụp xuống
Nguồn: Elements of Morphology, National Human Genome Research Institute
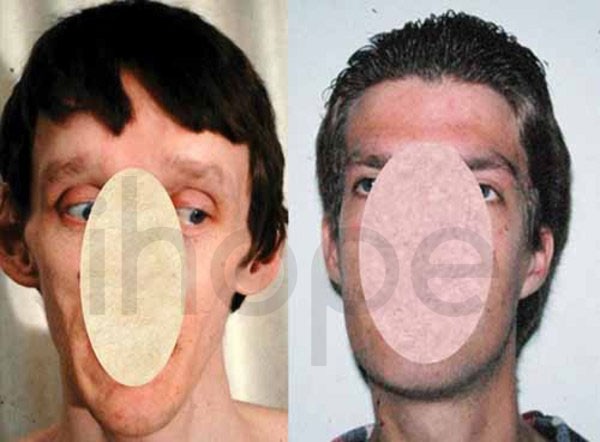
Ảnh: Mặt dài và hẹpNguồn: U.S. National Library of Medicine
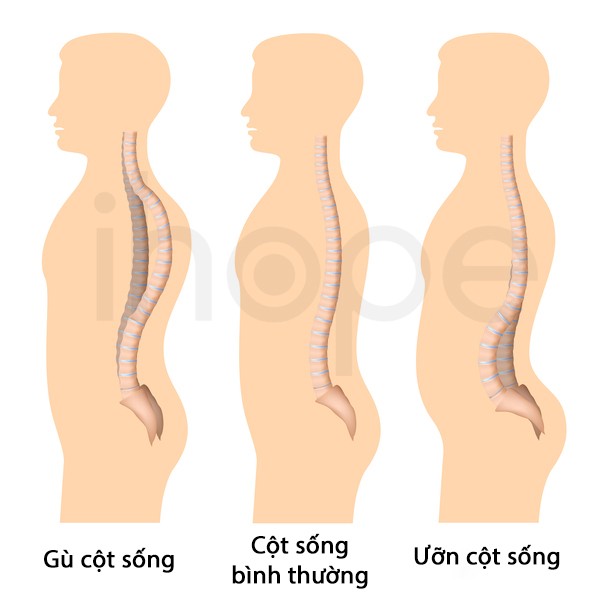
Ảnh: So sánh cột sống bình thường với gù cột sống và ưỡn cột sống
Nguồn: Alila Medical Media/Shutterstock.com
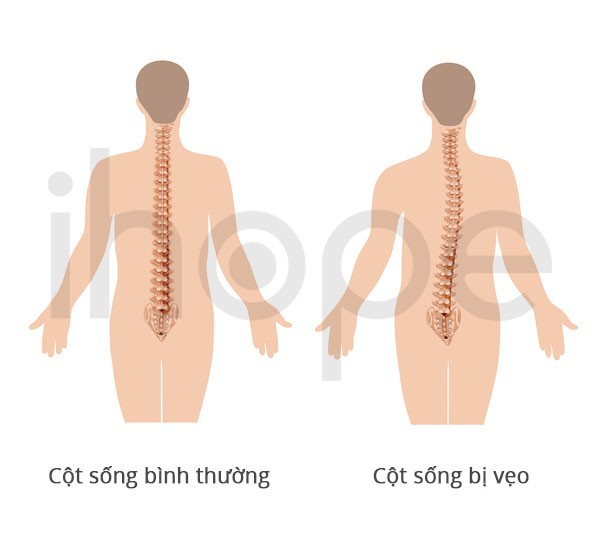
Ảnh: Cột sống bình thường và cột sống bị vẹo
Nguồn: U.S. National Library of Medicine
Độ phổ biến
Bệnh cơ mũ rất hiếm gặp. Hiện nay, người ta đã ghi nhận một số trường hợp nhưng chưa có thống kê cụ thể tỉ lệ mắc bệnh.
Nguyên nhân
Đột biến gen ACTA1, TPM2, hoặc TPM3 gây bệnh cơ mũ. Những gen này cung cấp hướng dẫn tạo ra protein có chức năng quan trọng đối với cơ xương.
Nguồn: Designua/Shutterstock.com
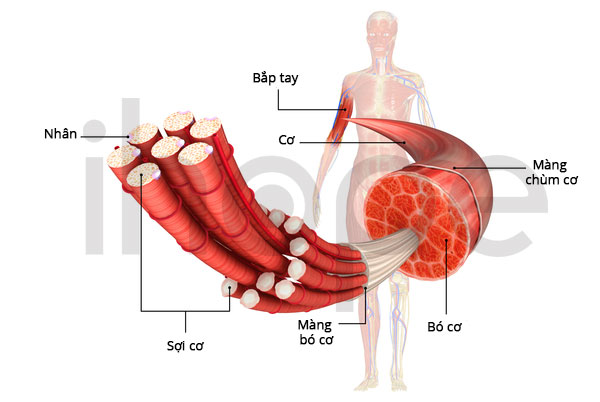
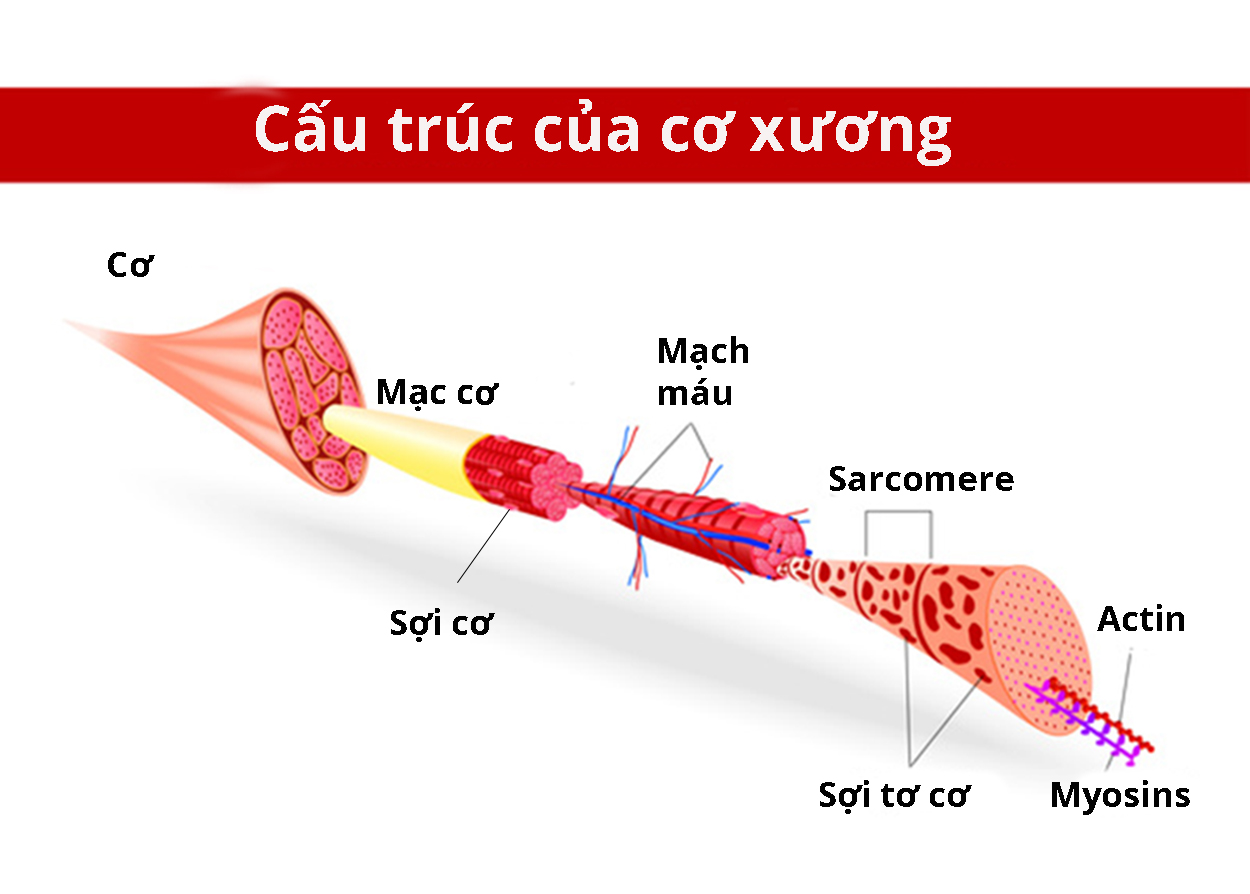
Gen ACTA1 cung cấp hướng dẫn tạo ra protein alpha (α)-actin trong xương. Protein này thuộc họ protein actin, chúng có chức năng quan trọng đối với quá trình di chuyển của tế bào và co cơ. Thành phần chính của bó cơ bao gồm sợi mỏng cấu tạo từ protein actin và sợi dày cấu tạo từ protein myosin.

Nguồn: Journal of Muscle Research and Cell Motility
Trong quá trình co cơ, sợi mỏng liên kết, chồng lên và tách khỏi sợi dày, chúng trượt lên nhau làm cơ co lại. Đột biến gen ACTA1 tạo ra protein bất thường, do đó quá trình hình thành các sợi mỏng bị ảnh hưởng. Sợi mỏng sắp xếp không đúng vị trí là nguyên nhân khiến tế bào cơ của người bệnh có cấu trúc dạng mũ.
Nguồn: Lecturio
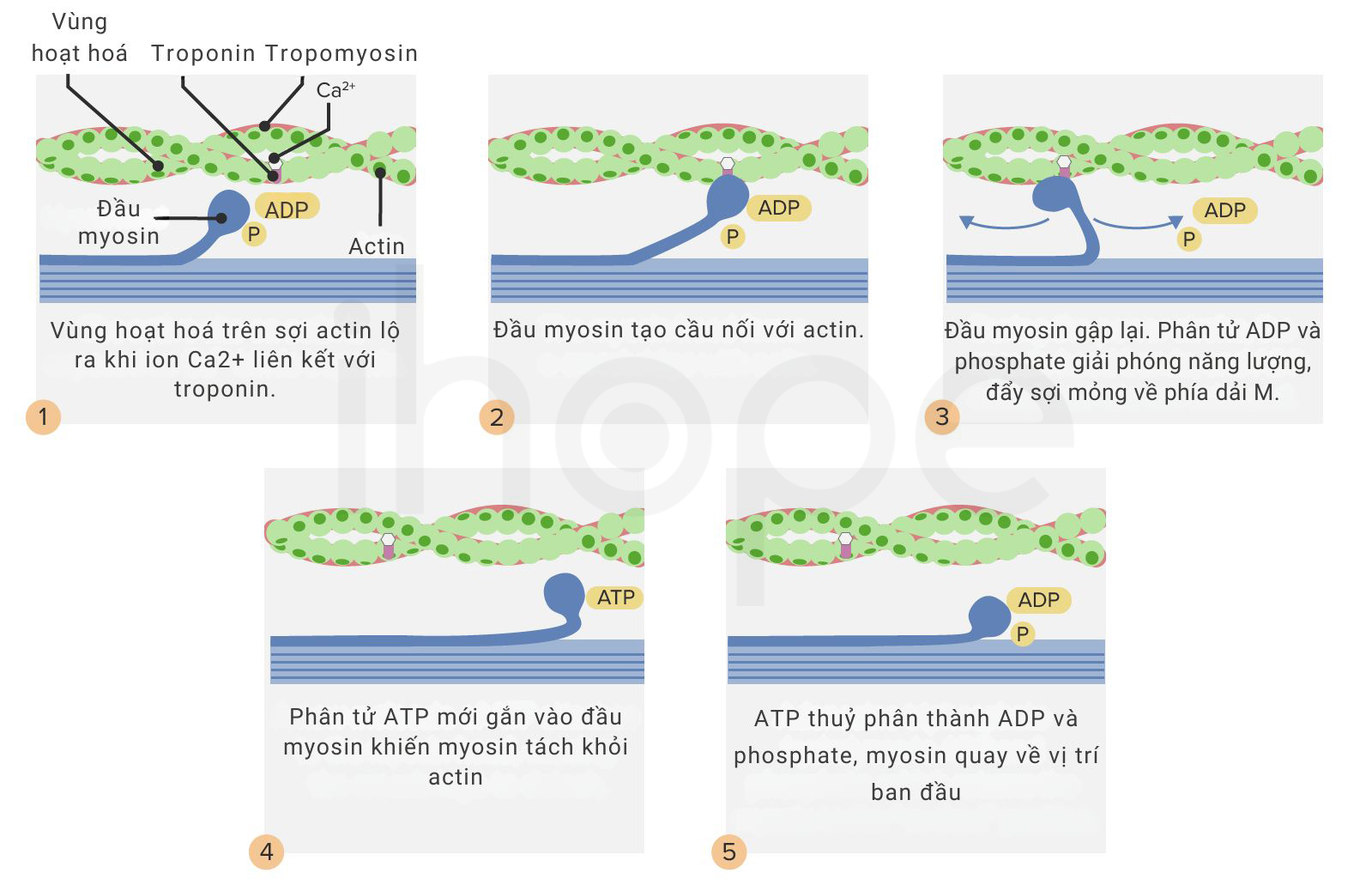
Gen TPM2 và TPM3 lần lượt cung cấp hướng dẫn tạo ra protein beta (β)-tropomyosin và alpha (α)-tropomyosin thuộc họ protein tropomyosin. Những protein này điều hoà hoạt động co cơ bằng cách liên kết với actin nhằm che vị trí liên kết giữa actin và myosin, do đó tương tác giữa chúng được kiểm soát. Đột biến gen TPM2 và TPM3 có thể ngăn cản liên kết giữa sợi mỏng với sợi dày, vì vậy hoạt động co cơ bị hưởng và cơ cũng yếu đi. Tuy nhiên, người ta chưa rõ cơ chế gây bệnh cụ thể của những đột biến này.
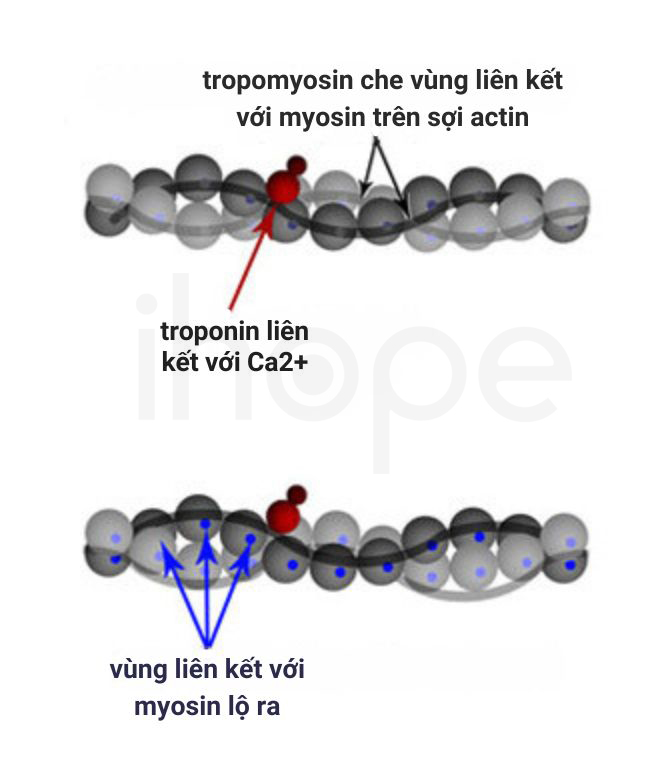
Nguồn: Nature Education
Chẩn đoán
Bác sĩ có thể chẩn đoán bệnh cơ mũ bằng cách sinh thiết và nhuộm mô, chụp cộng hưởng từ (MRI), xét nghiệm di truyền.
Sinh thiết và nhuộm mô
Sinh thiết và nhuộm mô là phương pháp cơ bản để chẩn đoán các bệnh cơ bẩm sinh. Sau khi sinh thiết, mẫu mô được quan sát dưới kính hiển vi quang học hoặc điện tử để phân tích mô học, hoá mô, hoá mô miễn dịch và siêu cấu trúc. Cấu trúc mũ thường xuất hiện tại vùng rìa và phân cách rõ ràng với phần còn lại của tế bào cơ.
Một số phương pháp nhuộm mô bao gồm:
- Nhuộm H&E
- Nhuộm NADH-TR
- Nhuộm actin và α-actinin
Khi quan sát mẫu mô sinh thiết bằng kính hiển vi điện tử, bác sĩ có thể phát hiện các cấu trúc mũ tập trung thành cụm tại vùng rìa sợi cơ. Những sợi cơ này sắp xếp theo nhiều hướng khác nhau và thường có dải protein đường Z ngăn cách giữa hai đơn vị co cơ.
Chụp cộng hưởng từ (MRI)
Kĩ thuật MRI sử dụng sóng vô tuyến và từ trường mạnh nhằm tạo ra hình ảnh chi tiết các mô trong cơ thể. Bác sĩ có thể chẩn đoán bệnh cơ mũ dựa vào dấu hiệu trên phim chụp MRI cùng với biểu hiện lâm sàng.
Xét nghiệm di truyền
Bác sĩ có thể chỉ định xét nghiệm di truyền nhằm phát hiện đột biến gen ACTA1, TPM2 hoặc TPM3 gây bệnh cơ mũ.
Điều trị
Hiện nay, chưa có phương pháp điều trị hoàn toàn bệnh cơ mũ. Đối với các bệnh lí về cơ, bác sĩ thường chỉ định trị liệu vật lí, trị liệu hoạt động cũng như một số bài tập khác nhằm giảm yếu cơ và cải thiện triệu chứng.
Dạng di truyền
Bệnh cơ mũ di truyền theo kiểu trội trên nhiễm sắc thể thường. Do đó, một bản sao của gen trong mỗi tế bào đủ để gây ra bệnh. Người bệnh thường được di truyền từ cha hoặc mẹ mắc bệnh. Một số ít trường hợp là do đột biến mới (denovo) xuất hiện trong quá trình hình thành tế bào sinh sản (trứng, tinh trùng) hoặc quá trình phát triển phôi sớm.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Bệnh cơ mũ di truyền trội trên nhiễm sắc thể thường, nếu cha hoặc mẹ mắc bệnh, con sinh ra sẽ có 50% khả năng di truyền bệnh. Do đó, để bảo đảm 100% khả năng con không bị bệnh, cha mẹ có thể chọn phương pháp thụ tinh nhân tạo IVF và sàng lọc phôi PGS/PGD. Các thành viên trong gia đình nên khám sức khỏe và tầm soát bệnh định kì nếu có thành viên mắc bệnh. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh.
Các tên gọi khác
- Cap disease
- Congenital myopathy with caps
References
- Genetic and Rare Diseases Information Center. Cap myopathy. Retrieved August 06, 2024 from https://rarediseases.info.nih.gov/diseases/11915/index
- Catalog of Genes and Diseases from OMIM. CONGENITAL MYOPATHY 4B, AUTOSOMAL RECESSIVE; CMYO4B. Retrieved August 06, 2024 from https://omim.org/entry/609284
- U.S National Library of Medicine. Cap myopathy. Retrieved August 06, 2024 from https://medlineplus.gov/genetics/condition/cap-myopathy/
- MalaCards. Cap Myopathy. Retrieved August 06, 2024 from https://www.malacards.org/card/cap_myopathy
- National Institute of Health. Congenital myopathies: clinical phenotypes and new diagnostic tools. Retrieved August 06, 2024 from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5688763/
- National Organization for Rare Disorders. Cap myopathy. Retrieved August 06, 2024 from https://rarediseases.org/gard-rare-disease/cap-myopathy/
- Orphanet. Cap myopathy. Retrieved August 06, 2024 from https://www.orpha.net/en/disease/detail/171881
- Radiopaedia. Myopathy. Retrieved August 06, 2024 from https://radiopaedia.org/articles/Myopathy





