
Hội chứng rối loạn chức năng đa ti thể (multiple mitochondrial dysfuntions syndrome) là bệnh di truyền liên quan đến tình trạng giảm chức năng của ti thể—bào quan sản xuất năng lượng cho cơ thể. Bình thường, một số rối loạn chức năng nhất định chỉ ảnh hưởng đến một bước trong quá trình sản xuất năng lượng nhưng hội chứng rối loạn chức năng đa ti thể ảnh hưởng đến nhiều giai đoạn trong quá trình này.
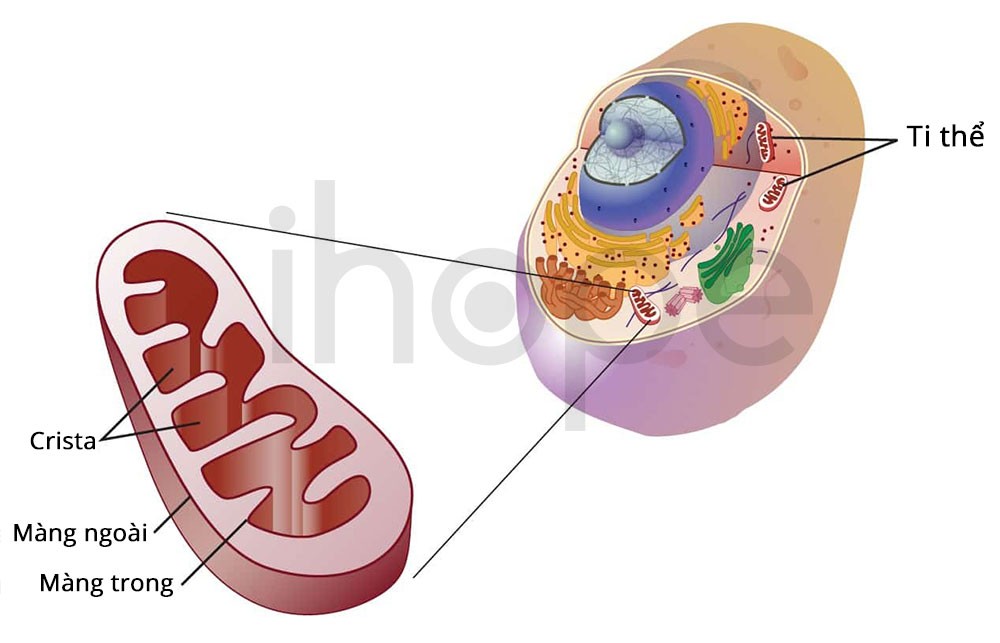
Nguồn: National Human Genome Research Institute
Biểu hiện lâm sàng
Triệu chứng của hội chứng rối loạn chức năng đa ti thể thường khởi phát sớm và nghiêm trọng nên người bệnh thường không sống qua giai đoạn trẻ nhỏ.
Trẻ sơ sinh mắc bệnh có các biểu hiện phổ biến sau:
- Rối loạn chức năng não nghiêm trọng
- Trương lực cơ yếu
- Co giật
- Chậm phát triển tâm thần vận động
Ngoài ra, trẻ bệnh thường có dấu hiệu suy dinh dưỡng. Đồng thời, bệnh còn khiến cơ thể người bệnh tích tụ axit lactic dẫn đến nguy cơ đe dọa tính mạng.
Một số biểu hiện khác của bệnh bao gồm:
- Tăng glycine trong máu
- Tăng đường huyết
- Tăng huyết áp phổi
- Cơ tim yếu
Nguồn: U.S. National Library of Medicine
Độ phổ biến
Hiện nay, tỉ lệ mắc hội chứng rối loạn chức năng đa ti thể chưa được xác định. Bệnh này rất hiếm gặp, nó ảnh hưởng đến 1/5.000 người trên toàn thế giới.
Nguyên nhân
Đột biến gen NFU1 hoặc BOLA3 gây ra hội chứng rối loạn chức năng đa ti thể. Hai gen này cung cấp hướng dẫn tạo ra protein cho quá trình tạo ra các cụm phân tử sắt-lưu huỳnh (Fe-S) hoặc gắn những cụm phân tử này vào protein khác. Một số protein cần kết hợp thêm các cụm sắt-lưu huỳnh để hoạt động bình thường.
Protein NFU-1 và BOLA3 giữ vai trò thiết yếu trong ti thể. Trong ti thể, nhiều protein có chức năng thúc đẩy các phản ứng hóa học để chuyển hóa năng lượng từ thức ăn thành dạng đơn giản cho tế bào sử dụng. Trong số đó có nhiều protein cần phải kết hợp với cụm sắt-lưu huỳnh để hoạt động.
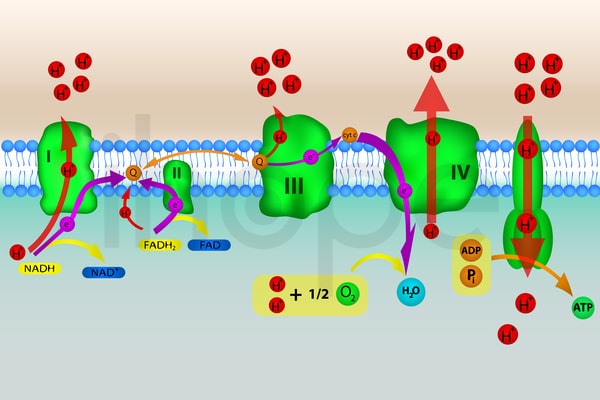
Nguồn: extender_01/Shutterstock.com
Các phức hợp protein cần kết hợp với cụm sắt-lưu huỳnh bao gồm:
- Phức hợp I
- Phức hợp II
- Phức hợp III
Ngoài ra, một protein khác trong ti thể cũng cần liên kết với cụm sắt-lưu huỳnh để hoạt động. Protein này biến đổi thêm những protein khác để hỗ trợ chuyển đổi năng lượng trong ti thể bao gồm phức hợp pyruvate dehydrogenase và phức hợp alpha-ketoglutarate dehydrogenase (tên gọi khác là phức hợp oxoglutarate dehydrogenase). Quá trình biến đổi này rất quan trọng trong hệ thống phân giải glycine. Hệ thống này gồm những protein tham gia phân giải glycine khi nồng độ axit amin này trong máu tăng cao.
Đột biến gen NFU1 và BOLA3 làm giảm hoặc loại bỏ quá trình tạo ra những protein tương ứng dẫn đến giảm quá trình hình thành cụm sắt-lưu huỳnh. Thiếu cụm sắt-lưu huỳnh sẽ ảnh hưởng đến chức năng của những protein liên quan đến quá trình tạo ra năng lượng và phân giải glycine. Hoạt động của những phức hợp I, II, III, phức hợp pyruvate dehydrogenase và phức hợp alpha-ketoglutarate dehydrogenase giảm sẽ làm tăng khả năng nhiễm toan lactic. Từ đó, đột biến có thể gây tử vong, bệnh não và dấu hiệu của hội chứng rối loạn chức năng đa ti thể. Trong một số trường hợp, người bệnh biểu hiện dấu hiệu tích lũy glycine do quá trình phân giải glycine giảm.
Chẩn đoán
Bác sĩ chẩn đoán hội chứng rối loạn chức năng đa ti thể dựa trên triệu chứng lâm sàng, kết quả từ những xét nghiệm sinh hóa và mô bệnh học.
Các xét nghiệm phổ biến gồm có:
- Xét nghiệm máu và nước tiểu để kiểm tra nồng độ axit lactic trong máu và nước tiểu. Dấu hiệu nhiễm toan lactic là một dấu hiệu nhận biết quan trọng. Tuy nhiên, triệu chứng này thường xuất hiện rõ ràng với những người bệnh nghiêm trọng, người bệnh nhẹ thường không có triệu chứng này.
- Xét nghiệm hình ảnh bao gồm chụp X-quang, chụp MRI.
- Kiểm tra thể chất nhằm đánh giá khả năng hô hấp của người bệnh. Người bệnh có ngưỡng kị khí thấp (quá trình trao đổi oxi giảm hoặc không hiệu quả) nên luôn phải tăng cường hô hấp.
- Điện cơ đồ (EMG) nhằm kiểm tra các thay đổi của điện cơ và phân loại thần kinh ngoại biên. Phần lớn người bệnh xuất hiện triệu chứng của bệnh cơ xương và bệnh thần kinh ngoại biên.
- Sinh thiết cơ để khảo sát hình thái và chức năng ti thể của người bệnh.
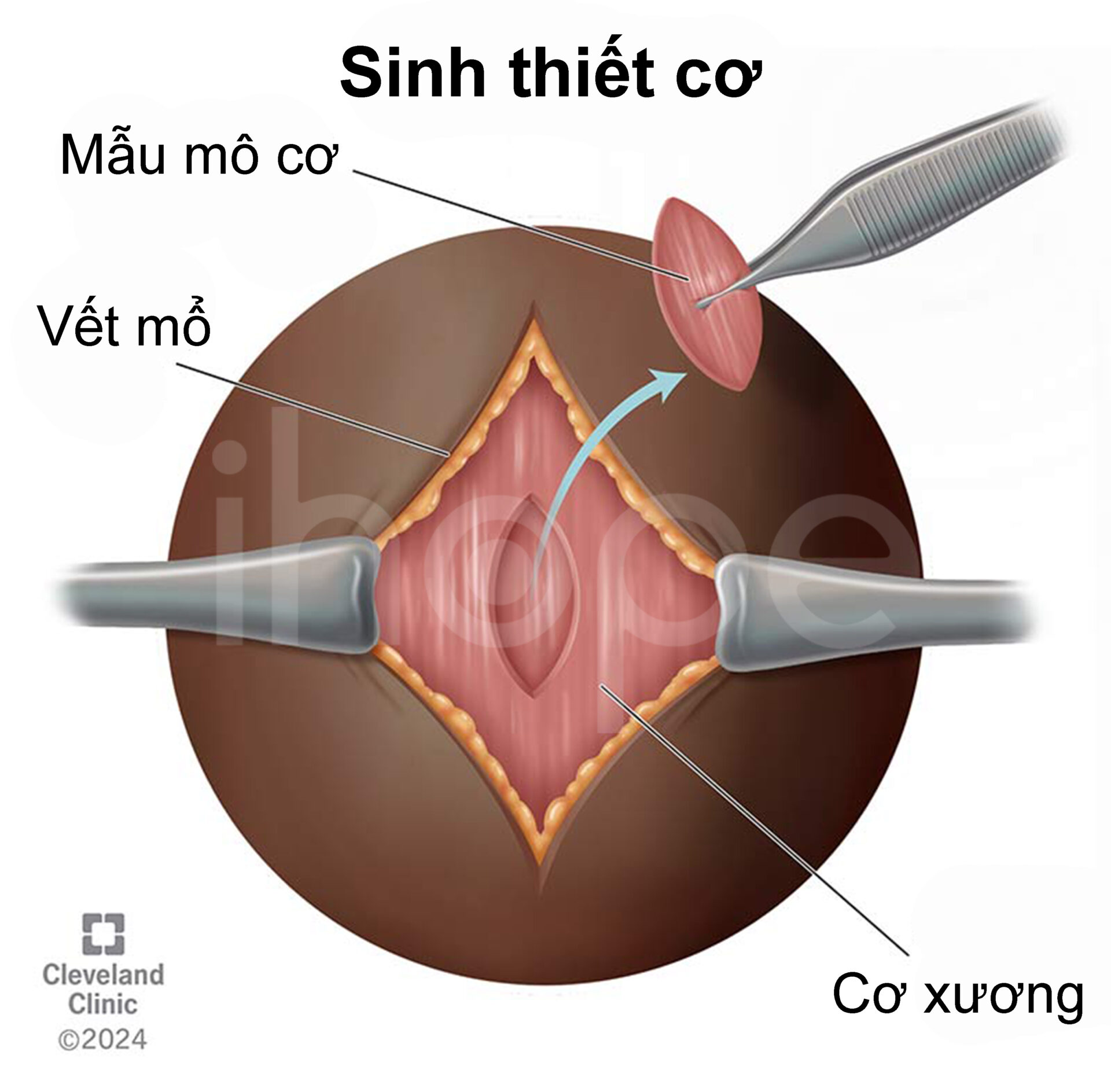
Nguồn: Cleveland Clinic
Ngoài ra, người bệnh có thể xét nghiệm di truyền để xác nhận kết quả chẩn đoán.
Điều trị
Hiện nay, chưa có liệu pháp điều trị hoàn toàn hội chứng rối loạn chức năng đa ti thể. Các liệu pháp tập trung giảm triệu chứng bệnh và cải thiện chất lượng cuộc sống người bệnh.
Các phương pháp phổ biến gồm có:
- Thuốc chống co giật
- Vitamin và thực phẩm chức năng như riboflacin, coenzyme Q10 và carnitine
- Thay đổi chế độ dinh dưỡng và tập luyện
- Vật lí trị liệu và trị liệu ngôn ngữ
Dạng di truyền
Hội chứng rối loạn chức năng đa ti thể di truyền theo kiểu lặn trên nhiễm sắc thể thường, nghĩa là cả hai bản sao của gen trong mỗi tế bào đều có đột biến. Cha mẹ của một cá nhân mắc bệnh lặn trên nhiễm sắc thể thường đều mang một bản sao của gen đột biến, nhưng họ thường không biểu hiện dấu hiệu và triệu chứng của bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Hội chứng rối loạn chức năng đa ti thể di truyền lặn do đột biến gen NFU1 hoặc BOLA3, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kỳ.
Các tên gọi khác
- MMDS
- Multiple mitochondrial dysfunction syndrome
References
- Genetic Testing Information. Multiple mitochondrial dysfunctions syndrome 1. Retrieved August 05, 2024 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C3276432/
- Genetic and Rare Diseases Information Center. Multiple mitochondrial dysfunctions syndrome. Retrieved August 05, 2024 from https://rarediseases.info.nih.gov/diseases/12632/index
- Catalog of Genes and Diseases from OMIM. MULTIPLE MITOCHONDRIAL DYSFUNCTIONS SYNDROME 1; MMDS1. Retrieved August 05, 2024 from https://omim.org/entry/605711
- U.S. National Library of Medicine. Multiple mitochondrial dysfunctions syndrome. Retrieved August 05, 2024 from https://medlineplus.gov/genetics/condition/multiple-mitochondrial-dysfunctions-syndrome/
- National Organization for Rare Disorders. Multiple mitochondrial dysfunctions syndrome. Retrieved August 05, 2024 from https://rarediseases.org/gard-rare-disease/multiple-mitochondrial-dysfunctions-syndrome/
- Orphanet. ISCA1-Related Multiple Mitochondrial Dysfunctions Syndrome. Retrieved August 05, 2024 from https://www.ncbi.nlm.nih.gov/books/NBK547304/
- Frontiers. Cavitating leukoencephalopathy with multiple mitochondrial dysfunction syndrome and NFU1 mutations. Retrieved August 05, 2024 from https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2014.00412/full
- MalaCards. Multiple Mitochondrial Dysfunctions Syndrome (MMDS). Retrieved August 05, 2024 from https://www.malacards.org/card/multiple_mitochondrial_dysfunctions_syndrome





