Tăng hồng cầu gia đình (familial erythrocytosis) là bệnh di truyền với biểu hiện tăng số lượng tế bào hồng cầu. Chức năng chính của hồng cầu là vận chuyển oxy từ phổi đến các mô và cơ quan khắp cơ thể.
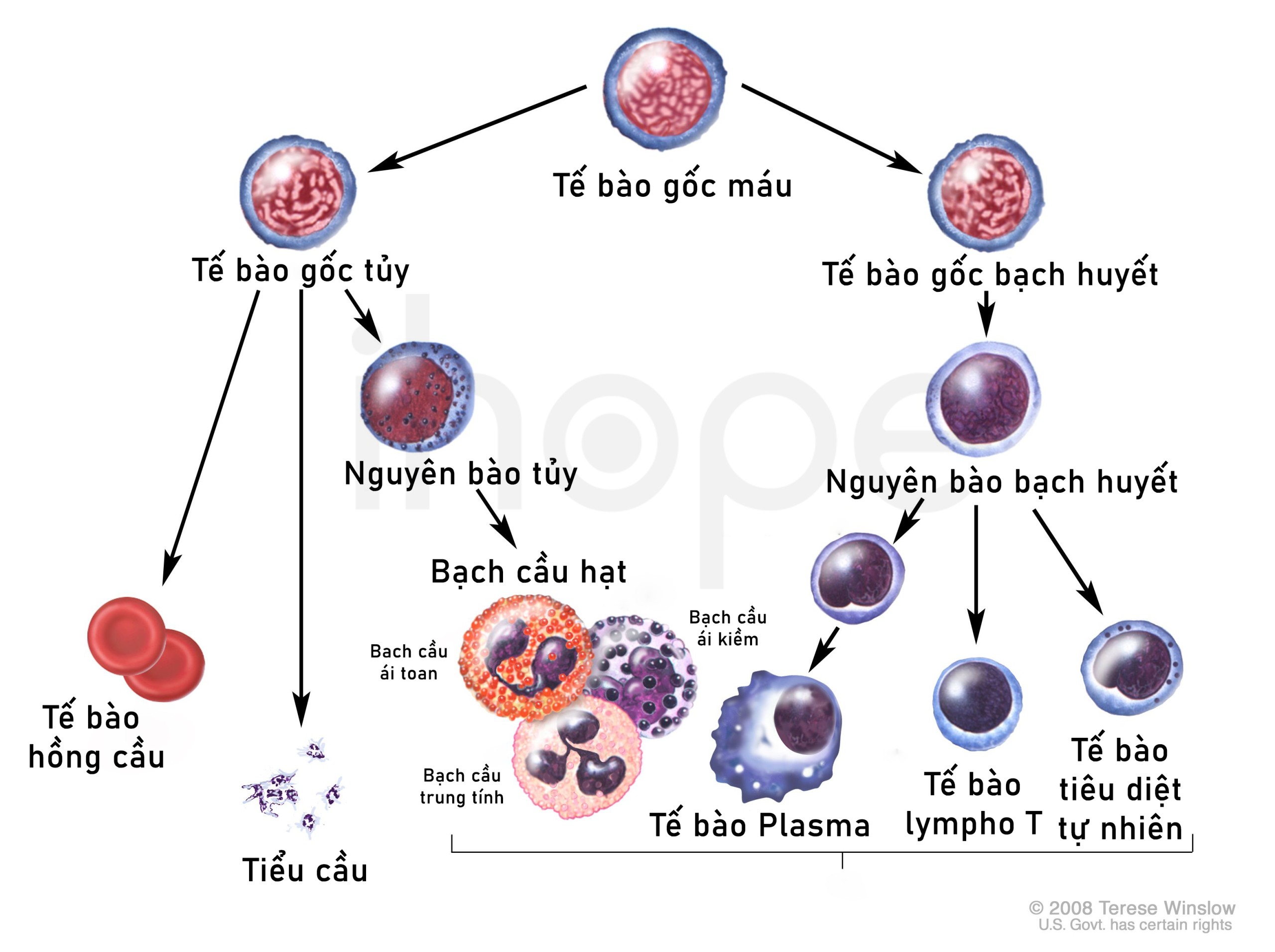
Nguồn:© 2008 Terese Winslow LLC for the National Cancer Institute
Biểu hiện lâm sàng
Các dấu hiệu của tăng hồng cầu gia đình bao gồm đau đầu, chóng mặt, khó thở và chảy máu cam. Tế bào hồng cầu dư thừa làm tăng nguy cơ phát triển cục máu đông bất thường, ngăn máu lưu thông qua động mạch và tĩnh mạch. Suy giảm tuần hoàn máu đến mô và cơ quan thiết yếu (tim, phổi hoặc não) gây ra biến chứng nguy hiểm như đau tim hay đột quỵ. Tuy nhiên, phần lớn trường hợp mắc bệnh dạng nhẹ hoặc không biểu hiện triệu chứng.
Độ phổ biến
Tăng hồng cầu gia đình hiếm gặp, do đó hiện nay tỷ lệ mắc bệnh vẫn chưa được thống kê cụ thể.
Nguyên nhân
Tăng hồng cầu gia đình do đột biến một trong các gen EPOR, VHL, EGLN1 hoặc EPAS1 gây ra.
Tăng hồng cầu gia đình được phân thành 4 loại dựa vào nguyên nhân di truyền như sau:
- Đột biến gen EPOR gây ra dạng ECYT1
- Đột biến gen VHL gây ra dạng ECYT2
- Đột biến gen EGLN1 gây ra dạng ECYT3
- Đột biến gen EPAS1 gây ra dạng ECTY4
Gen EPOR cung cấp hướng dẫn tạo ra protein thụ thể erythropoietin trên bề mặt tế bào tạo máu trong tủy xương. Erythropoietin là hormone có chức năng điều hoà quá trình sản xuất tế bào hồng cầu. Nó kết hợp với thụ thể theo cơ chế chìa khoá-ổ khoá nhằm kích hoạt tín hiệu hình thành tế bào hồng cầu. Đột biến gen EPOR khiến thụ thể erythropoietin được kích hoạt bất thường trong thời gian dài sau khi gắn vào erythropoietin. Thụ thể hoạt động quá mức nên nó truyền tín hiệu sản xuất tế bào hồng cầu ngay cả khi không cần thiết, dẫn đến dư thừa hồng cầu.
Các gen VHL, EGLN1 và EPAS1 tạo ra protein tham gia vào quá trình sản xuất hồng cầu bằng cách điều hoà erythropoietin. Protein tạo ra từ gen EPAS1 là thành phần của yếu tố cảm ứng khi thiếu oxy (hypoxia-inducible factor - HIF). Khi nồng độ oxy thấp hơn bình thường, HIF sẽ kích hoạt các gen giúp cơ thể thích nghi, bao gồm gen cung cấp hướng dẫn tạo ra erythropoietin. Protein tạo ra từ VHL và EGLN1 gián tiếp điều hoà erythropoietin bằng cách kiểm soát lượng HIF. Đột biến một trong các gen VHL, EGLN1 hoặc EPAS1 dẫn đến mất kiểm soát quá trình sản xuất hồng cầu.
Các nhà nghiên cứu đã mô tả dạng tăng hồng cầu mắc phải khi ở trên cao quá lâu, bệnh phổi, bệnh tim mãn tính, thở chậm hoặc ngưng thở khi ngủ, một số loại u. Một dạng khác của tăng hồng cầu mắc phải là bệnh đa hồng cầu nguyên phát do đột biến soma trong các gen liên quan đến quá trình sản xuất hồng cầu. Một số trường hợp chưa xác định được nguyên nhân gây bệnh.
Chẩn đoán
Chẩn đoán tăng hồng cầu gia đình thông qua biểu hiện lâm sàng, tiền sử bệnh gia đình. Bác sĩ sẽ thực hiện khám lâm sàng, kiểm tra các dấu hiệu và triệu chứng của bệnh nhân, bao gồm đường huyết, huyết áp, kích thước gan và thận. Bệnh nhân cần thực hiện xét nghiệm máu đo nồng độ hemoglobin và hematocrit, erythropoietin. Xét nghiệm di truyền xác định các đột biến gen liên quan đến bệnh. Bệnh nhân có thể thực hiện chụp CT hoặc MRI nhằm loại trừ các bệnh khác có triệu chứng tương tự.
Điều trị
Một số liệu pháp giúp thuyên giảm các triệu chứng của bệnh. Người bệnh cần lấy máu định kỳ nhằm duy trì lượng tế bào hồng cầu mức an toàn. Bên cạnh đó, bác sĩ có thể chỉ định sử dụng thuốc erythropoietin cho tác dụng tương đương như lấy máu. Người bệnh nên hạn chế sử dụng các thực phẩm giàu sắt như thịt đỏ, gan, rau chân vịt… Hiện nay chưa có phương pháp điều trị hoàn toàn tăng hồng cầu gia đình. Do đó, người bệnh phải thăm khám định kỳ nhằm đánh giá hiệu quả điều trị và phòng ngừa biến chứng tiềm ẩn.
Dạng di truyền
Tăng hồng cầu gia đình có các kiểu di truyền khác nhau tùy thuộc vào gen đột biến.
Bệnh do đột biến gen EPOR, EGLN1 hoặc EPAS1 di truyền theo kiểu trội trên nhiễm sắc thể thường. Do đó, một bản sao của gen trong mỗi tế bào đủ để gây ra bệnh. Phần lớn người bệnh thừa hưởng gen đột biến từ ba hoặc mẹ bị bệnh.

Nguồn: U.S. National Library of Medicine
Bệnh do đột biến gen VHL di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh lặn trên nhiễm sắc thể thường sẽ có bố và mẹ mang một bản sao của gen đột biến, nhưng bố mẹ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Tăng hồng cầu gia đình có cơ chế di truyền phức tạp. Các thành viên trong gia đình nên đi khám sức khỏe và tầm soát bệnh định kỳ nếu có thành viên mắc bệnh. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khoẻ mạnh.
Trường hợp bệnh di truyền trội trên nhiễm sắc thể thường, nếu cha hoặc mẹ mắc bệnh, con sinh ra sẽ có 50% khả năng di truyền bệnh. Do đó, để đảm bảo 100% khả năng con không bị bệnh, cha mẹ có thể chọn phương pháp thụ tinh nhân tạo IVF và sàng lọc phôi PGS/PGD.
Trường hợp bệnh di truyền lặn, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con.
Các tên gọi khác
- Benign familial polycythemia
- Congenital erythrocytosis
- Familial polycythemia
- Hereditary erythrocytosis
- Primary familial polycythemia
References
- Genetic Testing Information. Chuvash polycythemia. Retrieved April 27, 2023 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C1837915/
- Genetic and Rare Diseases Information Center. Primary familial and congenital polycythemia. Retrieved April 27, 2023 from https://rarediseases.info.nih.gov/diseases/9843/primary-familial-and-congenital-polycythemia
- Catalog of Genes and Diseases from OMIM. ERYTHROCYTOSIS, FAMILIAL, 1. Retrieved April 27, 2023 from https://omim.org/entry/133100
- U.S National Library of Medicine. Familial erythrocytosis. Retrieved April 27, 2023 from https://medlineplus.gov/genetics/condition/familial-erythrocytosis/
- National Organization for Rare Disorders. Primary familial and congenital polycythemia. Retrieved April 27, 2023 from https://rarediseases.org/gard-rare-disease/primary-familial-and-congenital-polycythemia/
- Orphanet. Chuvash erythrocytosis. Retrieved April 27, 2023 from https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=GB&Expert=238557
- Cario H. Childhood polycythemias/erythrocytoses: classification, diagnosis, clinical presentation, and treatment. Ann Hematol. 2005 Mar;84(3):137-45. doi: 10.1007/s00277-004-0985-1
- Hodges VM, Rainey S, Lappin TR, Maxwell AP. Pathophysiology of anemia and erythrocytosis. Crit Rev Oncol Hematol. 2007 Nov;64(2):139-58. doi: 10.1016/j.critrevonc.2007.06.006
- Huang LJ, Shen YM, Bulut GB. Advances in understanding the pathogenesis of primary familial and congenital polycythaemia. Br J Haematol. 2010 Mar;148(6):844-52. doi: 10.1111/j.1365-2141.2009.08069.x