Thiếu enzyme phân huỷ lipid trong lysosome (lysosomal acid lipase deficiency) là bệnh di truyền hiếm gặp, nguyên nhân bắt nguồn từ đột biến gen LIPA trên nhiễm sắc thể 10. Bệnh ảnh hưởng đến quá trình chuyển hóa chất béo trong tế bào, dẫn đến tình trạng lipid tích tụ trong cơ thể.
Biểu hiện lâm sàng
Mỗi bệnh nhân biểu hiện các triệu chứng với mức độ nghiêm trọng khác nhau.
Thiếu enzyme phân huỷ lipid trong lysosome được chia thành hai dạng dựa vào thời gian khởi phát bệnh:
Dạng khởi phát sớm (bệnh Wolman)
Bệnh có thể khởi phát khi trẻ mới sinh ra và thường có tiên lượng xấu. Một số ít trường hợp có thể sống sót qua một tuổi.
Lipid tích tụ trong cơ thể thai nhi, dẫn đến các triệu chứng như:
- Gan to
- Lách to
- Thành ruột dày
- Hấp thu dinh dưỡng kém
- Suy dinh dưỡng
- Gan nhiễm mỡ
- Suy gan
- Tuyến thượng thận to
- Vôi hóa tuyến thượng thận
- Suy vỏ thượng thận
- Tiêu chảy
- Nôn mửa
- Mỡ trong phân
- Thiếu sắt
- Vàng da
Nguồn: Shutterstock
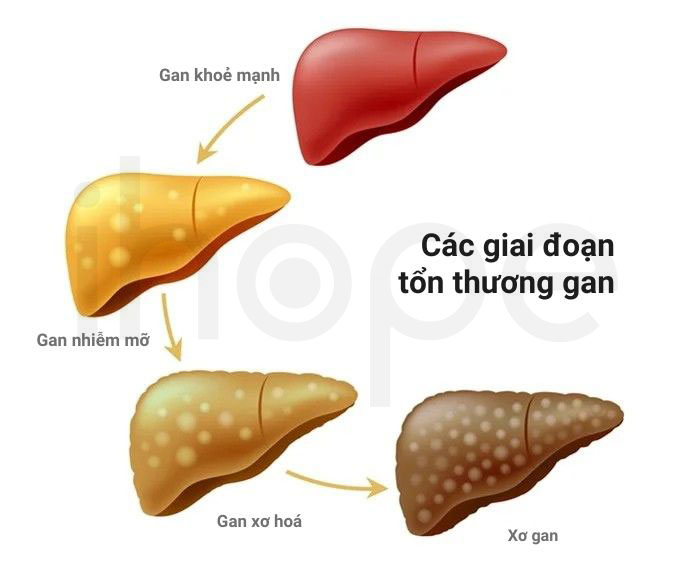
Dạng khởi phát muộn (bệnh tích tụ cholesterol ester)
Bệnh có biểu hiện lâm sàng tương tự dạng khởi phát sớm. Tuy nhiên, các triệu chứng bắt đầu xuất hiện khi trẻ lớn dần.
Ngoài ra, bệnh nhân còn biểu hiện một số triệu chứng khác như:
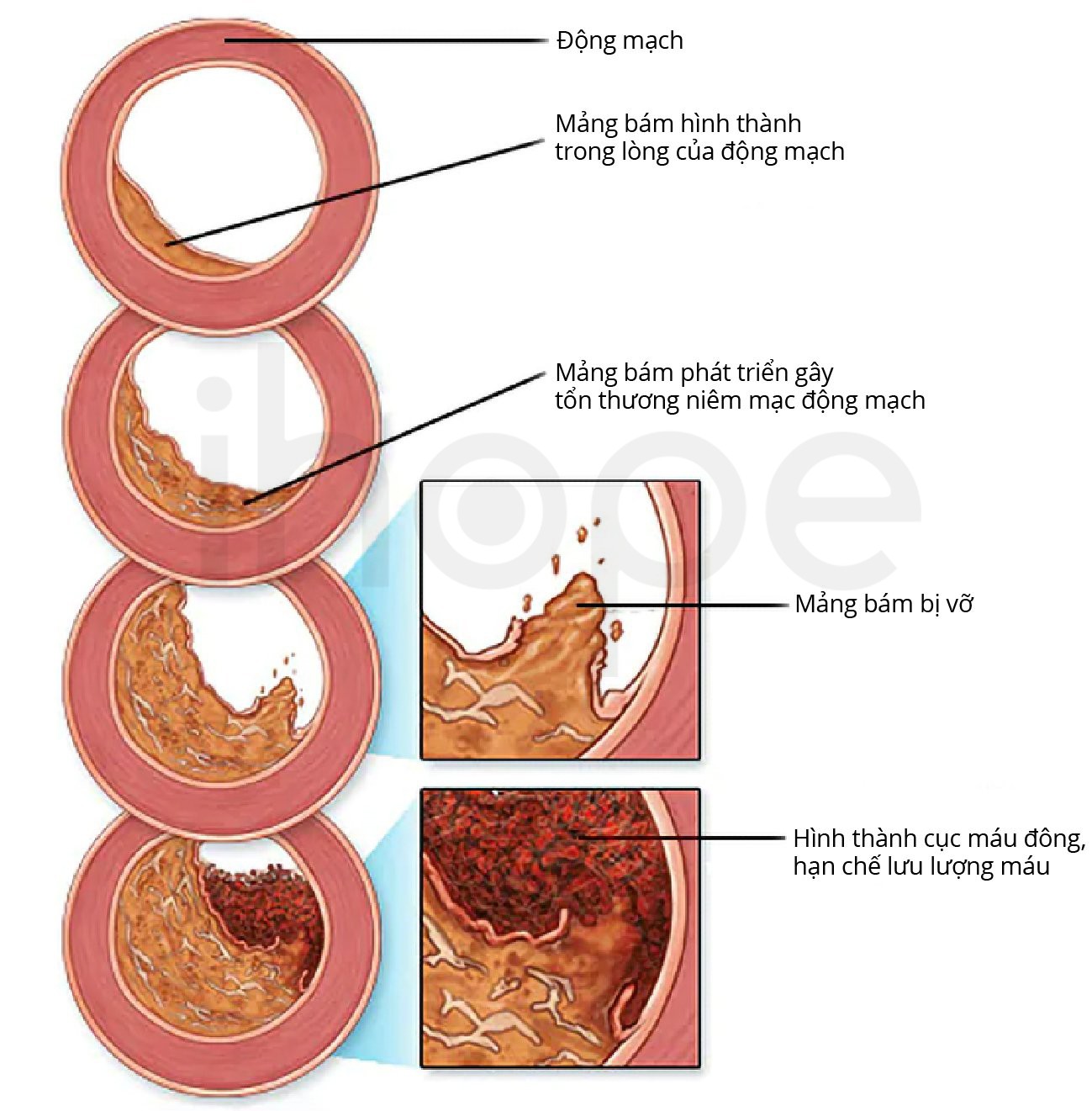
- Xơ vữa động mạch vành
- Giãn tĩnh mạch thực quản
- Xơ gan
- Sụt cân
- Chướng bụng
- Tăng huyết áp
- Xuất huyết đường tiêu hóa
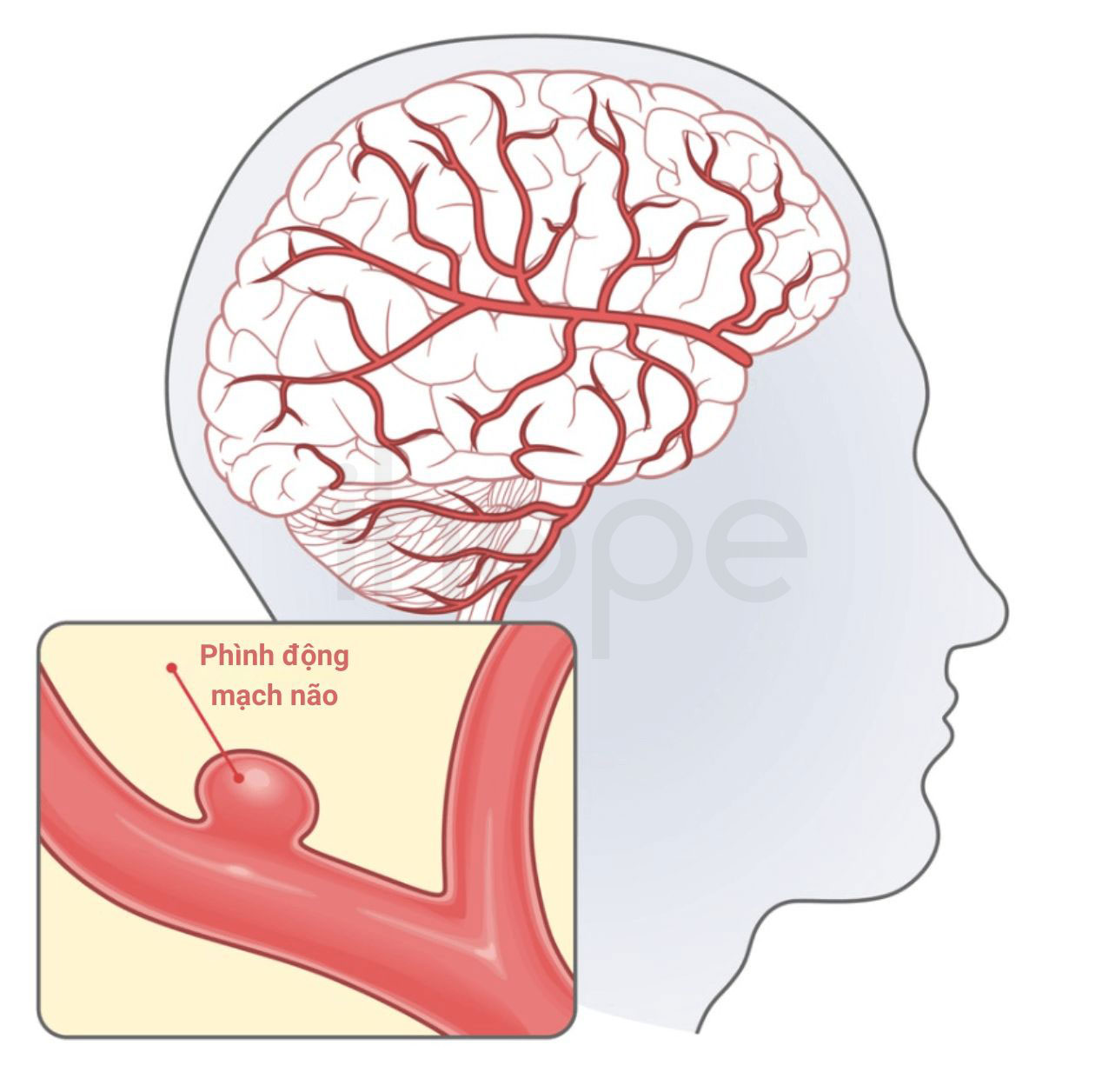
- Phình động mạch
- Giảm tiểu cầu
- Đột quỵ
Nguồn: Mayo Clinic
Nguồn: Supreme Vascular and Interventional Clinic
Độ phổ biến
Ước tính tỉ lệ mắc bệnh thiếu enzyme phân hủy lipid trong lysosome khoảng 1/177.000 người. Dạng khởi phát muộn phổ biến hơn dạng khởi phát sớm.
Nguyên nhân
Bệnh thiếu enzyme phân hủy lipid trong lysosome bắt nguồn từ đột biến gen LIPA trên nhiễm sắc thể 10. Gen LIPA cung cấp hướng dẫn tạo ra enzyme acid lipase. Enzyme này tham gia phân hủy lipid thành axit béo (tiền chất tạo cholesterol) trong bào quan lysosome nhằm tạo năng lượng cho cơ thể hoặc đào thải chất béo dư thừa qua gan.
Đột biến gen LIPA ảnh hướng đến cấu trúc và chức năng của enzyme acid lipase, do đó, quá trình phân hủy lipid bị gián đoạn. Lúc này, cơ thể không còn khả năng tổng hợp cholesterol từ acid béo nên đường tổng hợp cholesterol phụ được hoạt hóa rồi nồng độ cholesterol trong máu tăng lên. Lượng lipid dư thừa không được biến đổi tích tụ trong nhiều mô cơ quan đến mức gây ra các biểu hiện lâm sàng đặc trưng của bệnh. Mức độ nghiêm trọng của bệnh phụ thuộc vào lượng enzyme còn khả năng hoạt động bình thường.
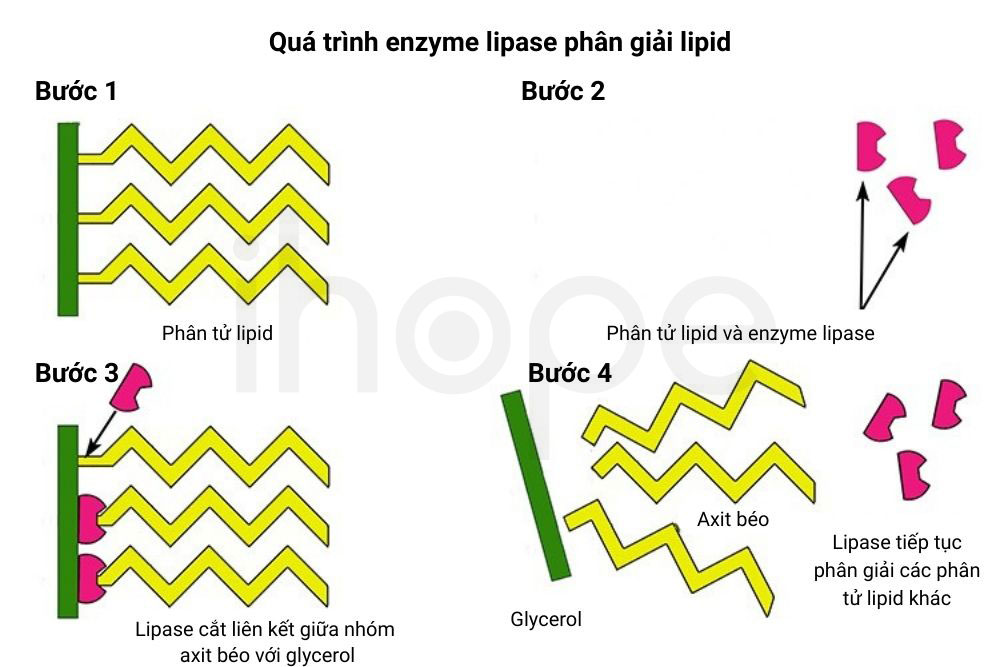
Nguồn: mammothmemory.net
Chẩn đoán
Thiếu enzyme phân hủy lipid trong lysosome được chẩn đoán dựa trên biểu hiện lâm sàng, bệnh sử gia đình kết hợp với kết quả xét nghiệm sinh hóa, mô, hình ảnh và di truyền.
Xét nghiệm sinh hóa
Bởi vì lipid tích tụ trong cơ thể, bệnh nhân thường có nồng độ lipid và lipoprotein trong máu bất thường. Xét nghiệm nồng độ lipid và lipoprotein trong máu có thể cho biết sớm các dấu hiệu của bệnh, từ đó bác sĩ đưa ra chẩn đoán ban đầu. Ngoài ra, enzyme acid lipase trong lysosome hoạt động kém là nguyên nhân gây bệnh thiếu enzyme phân hủy lipid trong lysosome. Do đó, bác sĩ tiến hành lấy mẫu máu hoặc da của bệnh nhân nhằm đánh giá hoạt tính của enzyme này. Ngoài ra, bệnh nhân có thể được chỉ định thêm các xét nghiệm chuyên sâu khác nhằm loại trừ những bệnh lý có biểu hiện tương tự.
Xét nghiệm mô
Bác sĩ sinh thiết gan và xử lí với hóa chất, sau đó quan sát mẫu mô dưới kính hiển vi nhằm phát hiện lipid tích tụ trong lysosome cũng như đánh giá mức độ tổn thương của mô gan. Đây là bước quan trọng để phân loại và xác định giai đoạn bệnh, từ đó phác đồ điều trị phù hợp được xây dựng.
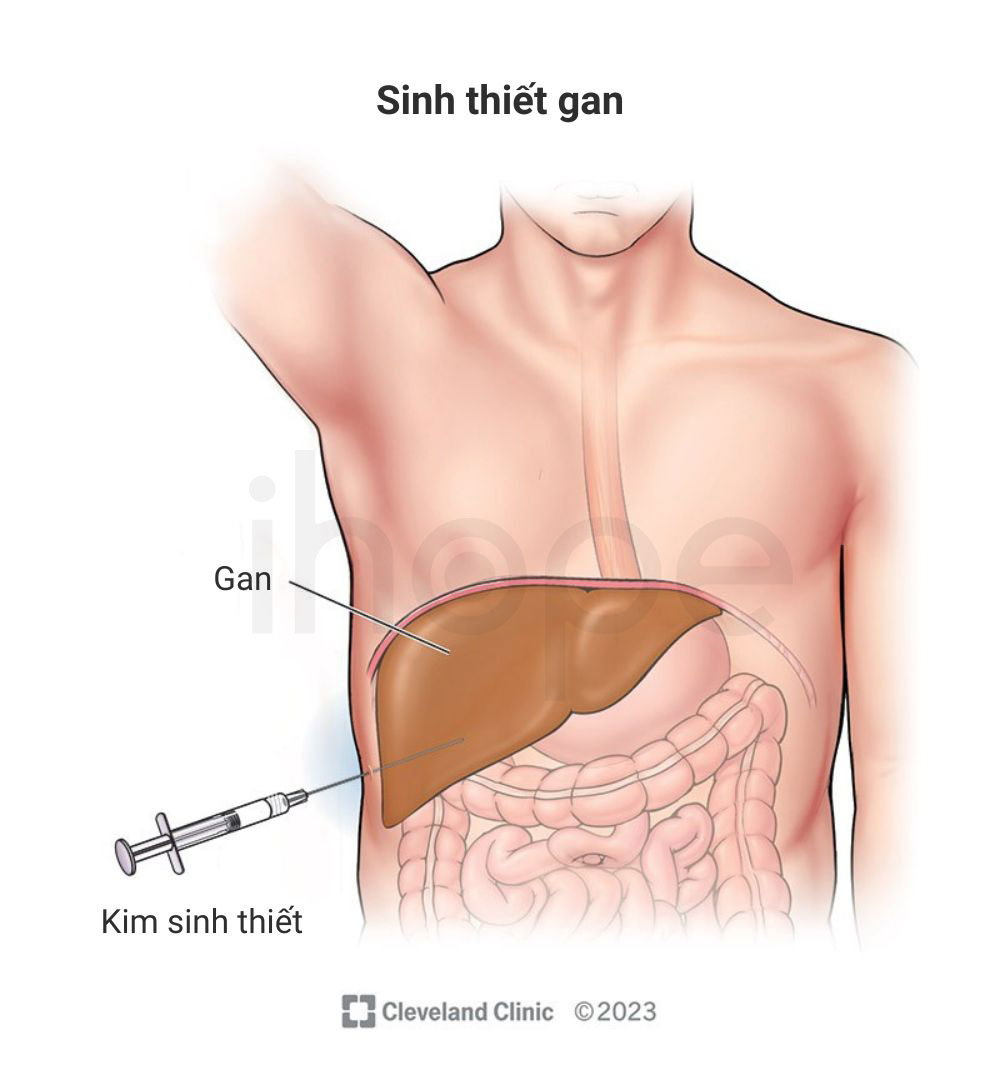
Nguồn: Cleveland Clinic
Chẩn đoán hình ảnh
Phương pháp chẩn đoán hình ảnh như chụp cộng hưởng từ (MRI), chụp cắt lớp (CT) hoặc siêu âm giúp bác sĩ phát hiện những bất thường trong cơ thể bệnh nhân.
Xét nghiệm di truyền
Bác sĩ thực hiện các xét nghiệm di truyền nhằm phát hiện đột biến gen gây bệnh, qua đó kết quả chẩn đoán được xác nhận.
Một số xét nghiệm di truyền nhằm phát hiện hội chứng bao gồm:
- Chuỗi polymerase thời gian thực (quantitative PCR)
- Khuếch đại đa trình tự bằng nhiều đoạn dò (MLPA)
- Giải trình tự gen (Sanger sequencing)
Điều trị
Hiện nay, chưa có phương pháp điều trị hoàn toàn bệnh thiếu enzyme phân hủy lipid trong lysosome. Các liệu pháp chủ yếu nhằm giảm nhẹ triệu chứng và cải thiện chất lượng đời sống người bệnh.
Phương pháp điều trị chủ yếu hiện nay là liệu pháp thay thế enzyme. Bệnh nhân được bổ sung enzyme nhân tạo nhằm thay thế lượng enzyme mất đi do đột biến gen. Ngoài ra, bệnh nhân cần áp dụng chế độ ăn ít chất béo và sử dụng thuốc giảm cholesterol trong máu như statin, cholestyramine, ezetimibe nhằm hỗ trợ điều trị bệnh. Bác sĩ có thể chỉ định ghép tế bào gốc tạo máu đối với trường hợp bệnh nhi mắc bệnh dạng khởi phát sớm.
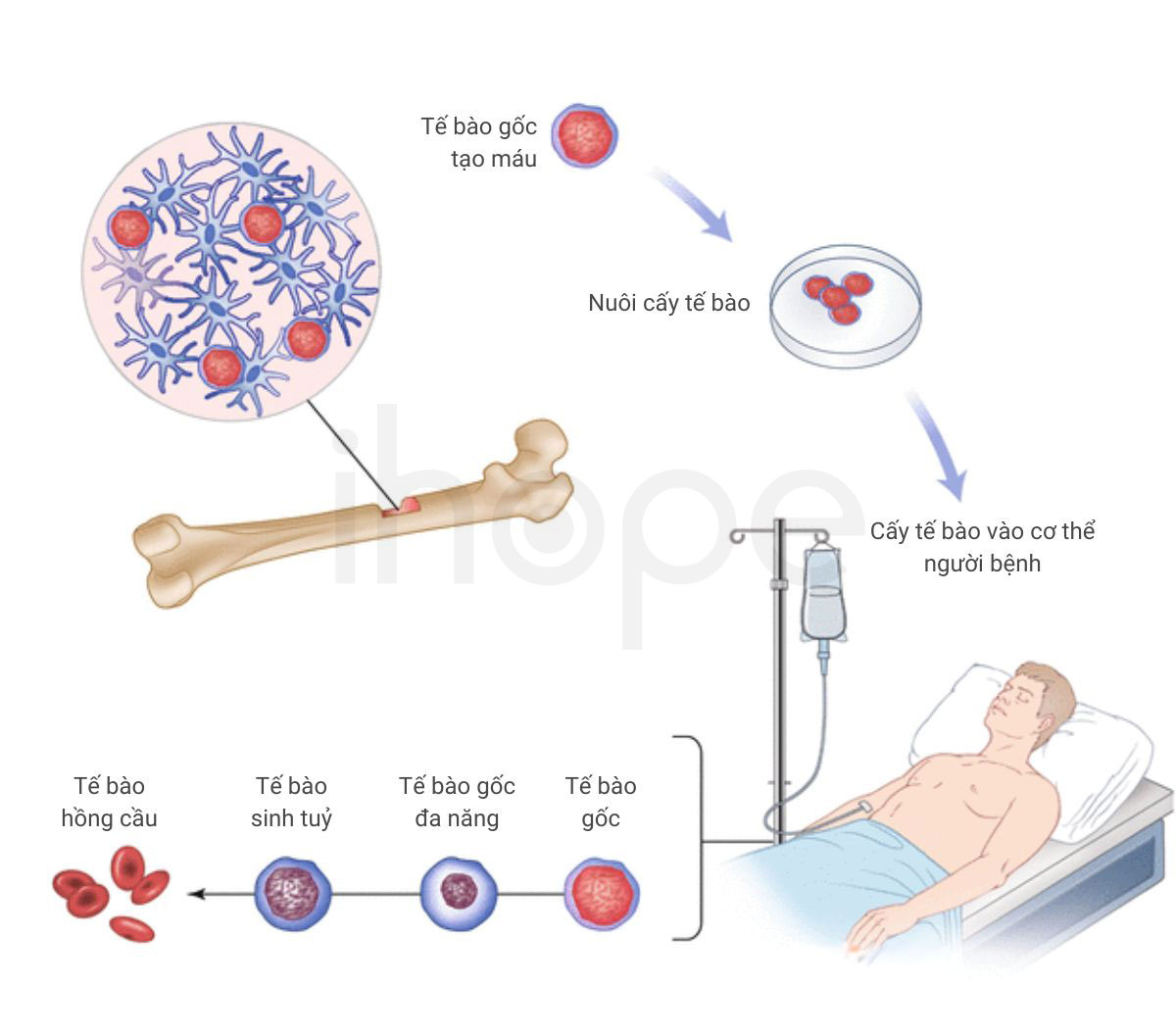
Nguồn: Springer
Dạng di truyền
Thiếu enzyme phân huỷ lipid trong lysosome di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh lặn trên nhiễm sắc thể thường sẽ có bố và mẹ mang một bản sao của gen đột biến, nhưng họ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Thiếu enzyme phân huỷ lipid trong lysosome di truyền theo kiểu lặn, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kì.
Các tên gọi khác
- Acid esterase deficiency
- Acid lipase deficiency
- Familial visceral xanthomatosis
- Familial xanthomatosis
- LAL deficiency
- LIPA deficiency
- Primary familial xanthomatosis
- Primary familial xanthomatosis with adrenal calcification
- Acid cholesterol ester hydrolase deficiency
- Cholesterol ester hydrolase deficiency storage disease
References
- National Library of Medicine. Wolman disease. Retrieved August 29, 2025 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0043208/
- Genetic and Rare Diseases Information Center. Lysosomal acid lipase deficiency. Retrieved August 29, 2025 from https://rarediseases.info.nih.gov/diseases/12097/index
- OMIM. CHOLESTERYL ESTER STORAGE DISEASE; CESD. Retrieved August 29, 2025 from https://omim.org/entry/278000
- MedlinePlus. Lysosomal acid lipase deficiency. Retrieved August 29, 2025 from https://medlineplus.gov/genetics/condition/lysosomal-acid-lipase-deficiency/
- GeneReviews. Lysosomal Acid Lipase Deficiency. Retrieved August 29, 2025 from https://www.ncbi.nlm.nih.gov/sites/books/NBK305870/
- Orphanet. Lysosomal acid lipase deficiency. Retrieved August 29, 2025 from https://www.orpha.net/en/disease/detail/275761