Bệnh cơ que nội nhân (intranuclear rod myopathy) chủ yếu ảnh hưởng đến cơ xương—cơ có chức năng vận động. Người bệnh có cấu trúc hình que bất thường trong nhân tế bào cơ. Biểu hiện chung của người bệnh gồm yếu cơ nghiêm trọng và giảm trương lực cơ toàn bộ cơ thể.
Nguồn: U.S. National Library of Medicine

Ảnh: Trương lực cơ giảm
Nguồn: Icahn School of Medicine at Mount Sinai
Biểu hiện lâm sàng
Các dấu hiệu và triệu chứng của bệnh cơ que nội nhân khởi phát trên trẻ sơ sinh gồm:
- Nuốt khó khăn
- Tiếng khóc yếu
- Khó kiểm soát chuyển động đầu
- Cơ thể mềm oặt
- Không thể tự di chuyển
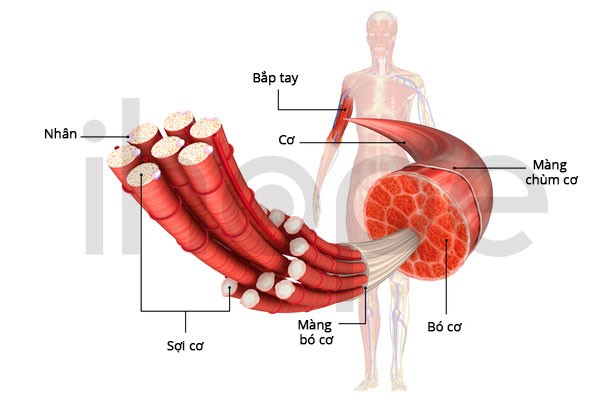
Yếu cơ cũng ảnh hưởng đến các cơ sử dụng để thở. Vì vậy, người bệnh có dấu hiệu thở nông, đặc biệt khi ngủ, dẫn đến cơ thể thiếu oxy và tích tụ carbon dioxide trong máu. Ngoài ra, nhiễm trùng đường hô hấp và khó thở có thể đe dọa đến tính mạng người bệnh. Đa phần người bệnh không sống qua giai đoạn sơ sinh do thường xuyên gặp vấn đề hô hấp. Nếu sống sót, người bệnh sẽ chậm phát triển các kĩ năng vận động như ngồi, bò, đứng và đi.
Nguồn: © 2006 Terese Winslow LLC for the National Cancer Institute
Độ phổ biến
Bệnh cơ que nội nhân rất hiếm gặp. Hiện nay, người ta đã xác định một số cá nhân mắc bệnh và chưa có thống kê cụ thể tỉ lệ mắc bệnh.
Nguyên nhân
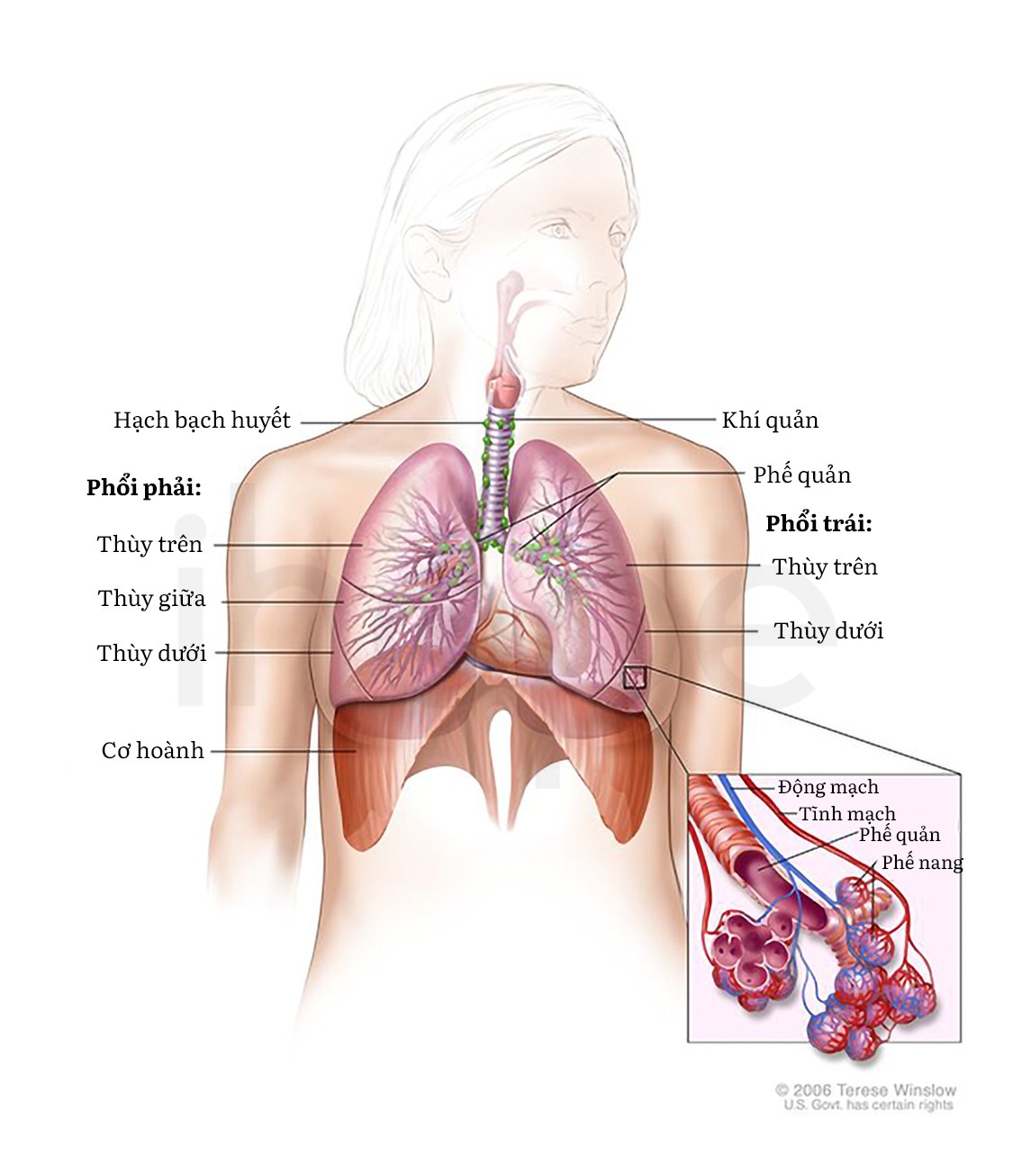
Đột biến gen ACTA1 gây ra bệnh cơ que nội nhân. Gen này cung cấp hướng dẫn tạo ra protein alpha (α)-actin xương thuộc họ protein actin. Protein actin có chức năng quan trọng đối với chuyển động của tế bào và hoạt động co cơ.
Nguồn: U.S National Library of Medicine
Protein alpha (α)-actin xương có chức năng quan trọng đối với cơ xương—cơ dùng để vận động. Cụ thể, protein này tham gia cấu tạo nên sarcomere trong tế bào cơ xương. Cấu trúc sarcomere bao gồm sợi mỏng tạo thành từ actin và sợi dày cấu tạo từ myosin. Trong quá trình co cơ, sợi mỏng liên kết, chồng lên và tách khỏi sợi dày, chúng trượt lên nhau làm cơ co lại.

Nguồn: U.S National Library of Medicine
Đột biến gen ACTA1 dẫn đến tình trạng actin dạng que tích tụ trong nhân của tế bào cơ. Đối với người bình thường, phần lớn actin hiện diện tại tế bào chất và một lượng nhỏ tồn tại trong nhân. Trường hợp người bệnh, người ta nhận thấy đột biến gen ACTA1 có thể ngăn cản quá trình vận chuyển actin giữa nhân và tế bào chất, do đó, actin tích tụ trong nhân rồi hình thành dạng que. Lượng actin trong các sợi cơ sẽ giảm đi tương ứng với actin tích tụ trong nhân tế bào cơ, vì vậy hoạt động co cơ không hiệu quả và gây ra triệu chứng yếu cơ cho người bệnh.
Mặt khác, một số người bệnh không mang đột biến gen ACTA1. Người ta chưa rõ nguyên nhân gây bệnh trong những trường hợp này.
Chẩn đoán
Bác sĩ chẩn đoán bệnh cơ que nội nhân dựa trên bài kiểm tra phản xạ, sức mạnh cơ kết hợp với các xét nghiệm chuyên biệt và bệnh sử gia đình.
Một số xét nghiệm phổ biến bao gồm:
- Xét nghiệm máu
- Điện cơ đồ
- Chụp cộng hưởng từ (MRI) cơ
- Sinh thiết cơ
Ngoài ra, bác sĩ có thể chỉ định bệnh nhân làm xét nghiệm di truyền để xác định đột biến gen ACTA1, qua đó kết quả chẩn đoán được xác nhận.
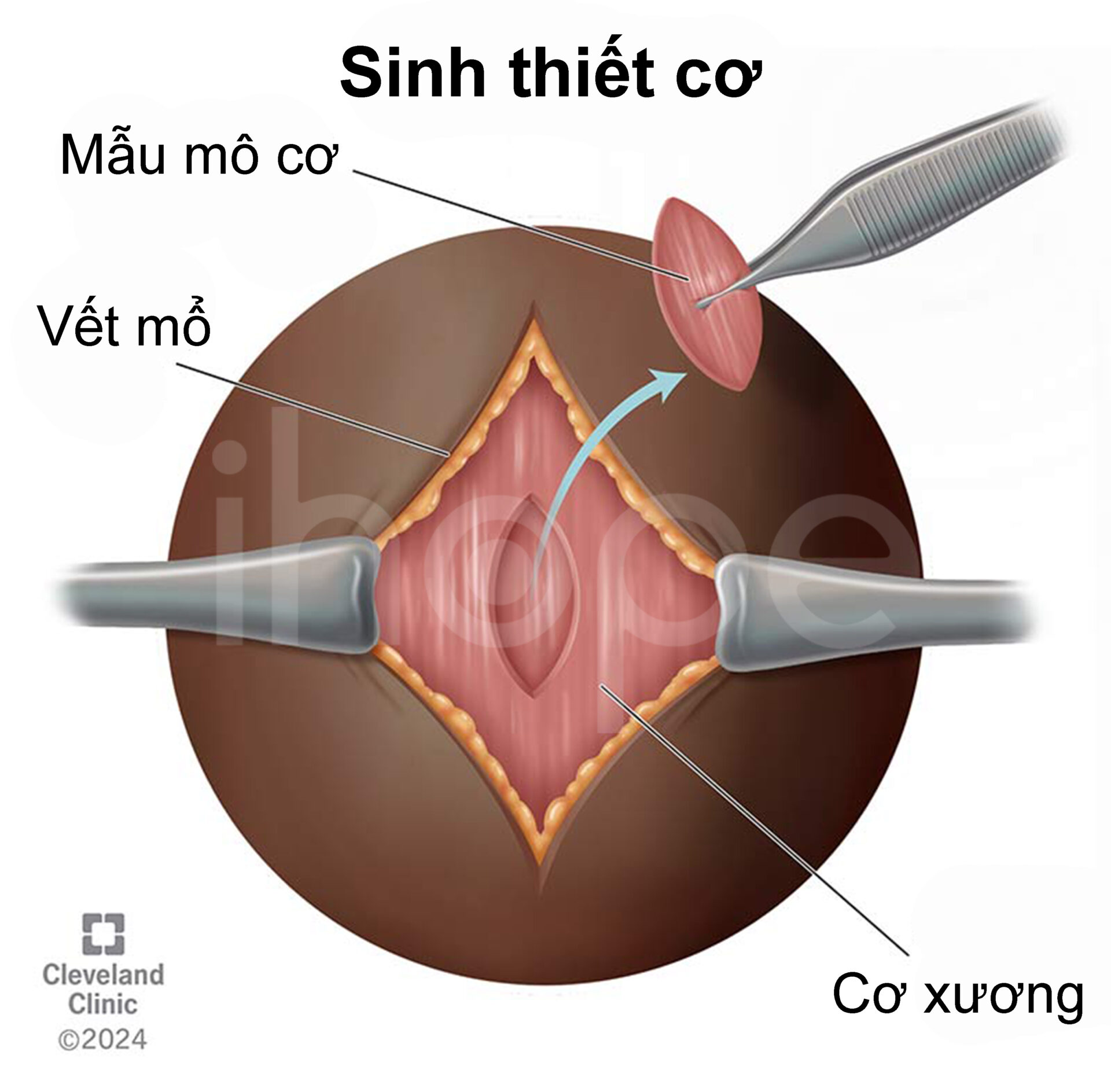
Ảnh: Sinh thiết cơ
Nguồn: Cleveland Clinic
Điều trị
Hiện nay, chưa có phương pháp điều trị hoàn toàn bệnh cơ que nội nhân. Các liệu pháp chủ yếu nhằm duy trì sức mạnh cơ, khả năng vận động và chuyển động của khớp thông qua vật lí trị liệu. Ngoài ra, bác sĩ cần thường xuyên theo dõi chức năng hô hấp của người bệnh. Trường hợp vẹo cột sống nghiêm trọng, người bệnh có thể thực hiện phẫu thuật chỉnh hình.
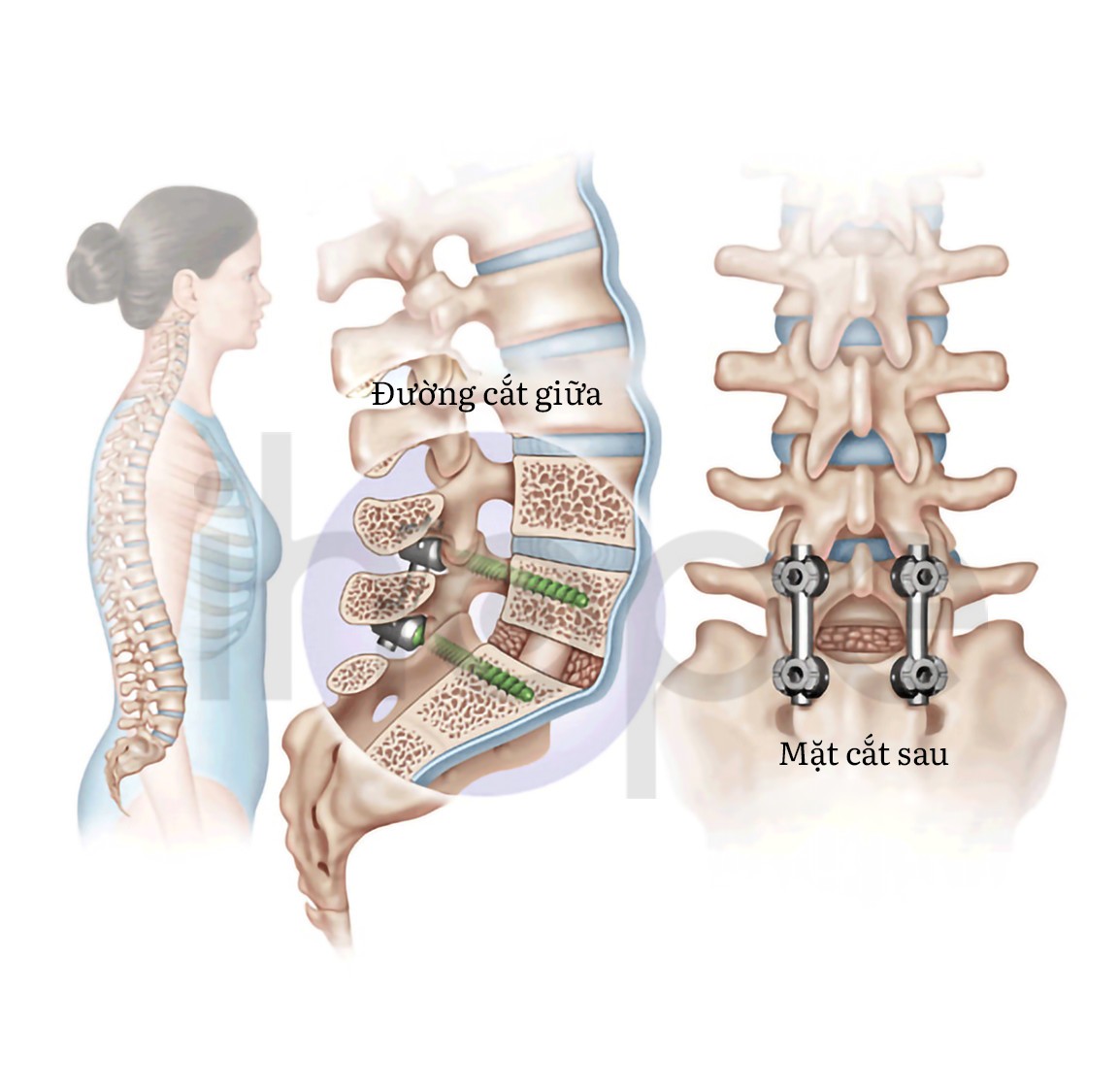
Nguồn: waynecheng.com
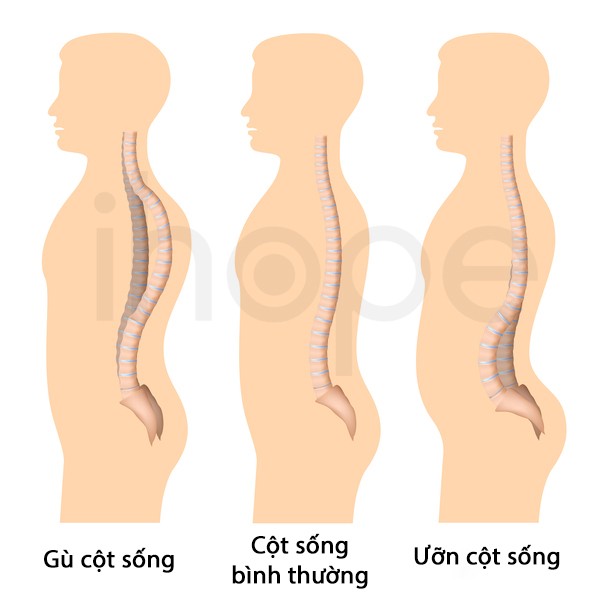
Ảnh: So sánh cột sống bình thường và cột sống cong, vẹo
Nguồn: Alila Medical Media/Shutterstock.com
Dạng di truyền
Bệnh cơ que nội nhân di truyền theo kiểu trội trên nhiễm sắc thể thường. Do đó, một bản sao của gen trong mỗi tế bào đủ để gây ra bệnh. Người bệnh thường được di truyền từ cha hoặc mẹ mắc bệnh. Một số ít trường hợp là do đột biến mới (denovo) xuất hiện trong quá trình hình thành tế bào sinh sản (trứng, tinh trùng) hoặc quá trình phát triển phôi sớm.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Bệnh cơ que nội nhân di truyền trội trên nhiễm sắc thể thường, nếu cha hoặc mẹ mắc bệnh, con sinh ra sẽ có 50% khả năng di truyền bệnh. Do đó, để bảo đảm 100% khả năng con không bị bệnh, cha mẹ có thể chọn phương pháp thụ tinh nhân tạo IVF và sàng lọc phôi PGS/PGD. Các thành viên trong gia đình nên khám sức khỏe và tầm soát bệnh định kì nếu có thành viên mắc bệnh. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh.
Các tên gọi khác
- Intranuclear nemaline rod myopathy
- Nemaline myopathy with exclusively intranuclear rods
References
- Genetic Testing Information. Actin accumulation myopathy. Retrieved September 15, 2024 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C3711389/
- Catalog of Genes and Diseases from OMIM. CONGENITAL MYOPATHY 2A, TYPICAL, AUTOSOMAL DOMINANT; CMYO2A. Retrieved September 15, 2024 from https://omim.org/entry/161800
- U.S National Library of Medicine. Intranuclear rod myopathy. Retrieved September 15, 2024 from https://medlineplus.gov/genetics/condition/intranuclear-rod-myopathy/
- Frontiers. Case report: A novel ACTA1 variant in a patient with nemaline rods and increased glycogen deposition. Retrieved September 15, 2024 from https://www.frontiersin.org/journals/neurology/articles/10.3389/fneur.2024.1340693/full
- MalaCards. Intranuclear Rod Myopathy. Retrieved September 15, 2024 from https://www.malacards.org/card/intranuclear_rod_myopathy
- National Institute of Health. Intranuclear rod myopathy. Retrieved September 15, 2024 from https://www.ncbi.nlm.nih.gov/medgen/777985
- Neurology Journals. Intranuclear rods in severe congenital nemaline myopathy. Retrieved September 15, 2024 from https://www.neurology.org/doi/10.1212/wnl.43.11.2372