Hội chứng Coats cộng (coats plus syndrome) là bệnh di truyền với các biểu hiện đặc trưng bao gồm rối loạn mắt (bệnh Coats) kết hợp với bất thường tại não, xương, hệ tiêu hóa và các bộ phận khác của cơ thể.
Biểu hiện lâm sàng
Bong võng mạc
Bệnh Coats ảnh hưởng đến võng mạc , khiến mạch máu tại võng mạc to bất thường và xoắn lại. Võng mạc là mô nằm phía sau mắt, chúng có chức năng phát hiện ánh sáng và màu sắc. Các mạch máu này rò rỉ dịch làm lớp võng mạc bong ra, vì vậy, người bệnh bị mất thị lực.
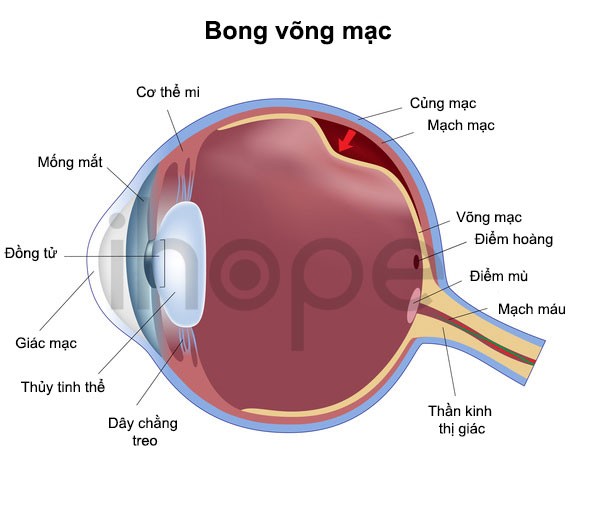
Nguồn: U.S. National Library of Medicine
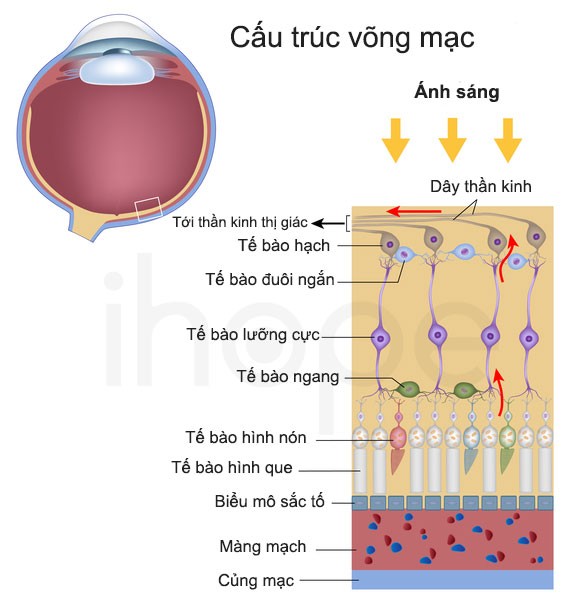
Ảnh: Cấu trúc võng mạc
Nguồn: U.S. National Library of Medicine
Não bất thường
Bệnh nhân mắc hội chứng Coats cộng cũng xuất hiện các bất thường về não như:
- Lắng đọng canxi (vôi hóa)
- Hình thành u nang
- Loạn dưỡng chất trắng
Những triệu chứng này tiến triển nghiêm trọng hơn theo thời gian, nên người bệnh bị chậm phát triển, rối loạn vận động, co giật và thiểu năng trí tuệ.
Hội chứng Coats cộng và hội chứng Labrune (leukoencephalopathy with calcifications and cysts—bệnh não chất trắng có vôi hóa và u nang) đều gây ra bất thường não giống nhau. Vì vậy, người ta thường gộp chung hai hội chứng này thành bệnh não võng mạc do vi mạch có vôi hóa và u nang (cerebroretinal microangiopathy with calcifications and cysts). Tuy nhiên, gần đây người ta phát hiện hội chứng Coats cộng và hội chứng Labrune có nguyên nhân di truyền khác nhau. Do đó, chúng được tách ra thành hai hội chứng riêng biệt thay vì gộp chung thành một nhóm bệnh.
Các biểu hiện khác
Hội chứng Coats cộng có thể gây loãng xương cũng như khiến xương trở nên giòn và dễ gãy. Bên cạnh đó, bệnh còn làm cơ thể thiếu tế bào hồng cầu, nên người bệnh có các biểu hiện như da xanh xao và mệt mỏi.
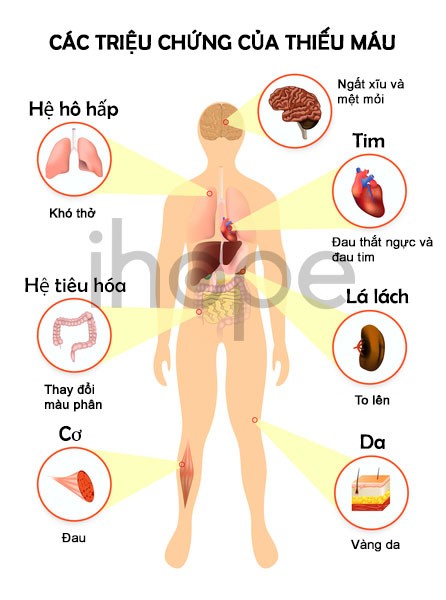
Nguồn: Designua/Shutterstock.com
Nghiêm trọng hơn, bệnh nhân có thể gặp những biến chứng nguy hiểm đe dọa tính mạng như:
- Suy gan
- Chảy máu đường tiêu hóa
- Tăng áp lực tĩnh mạch cửa (tĩnh mạch cung cấp máu cho gan
Các triệu chứng ít phổ biến của hội chứng Coats cộng bao gồm:
- Tóc thưa và bạc sớm
- Dị tật móng tay và móng chân
- Bất thường màu da (xuất hiện mảng da màu nâu nhạt )
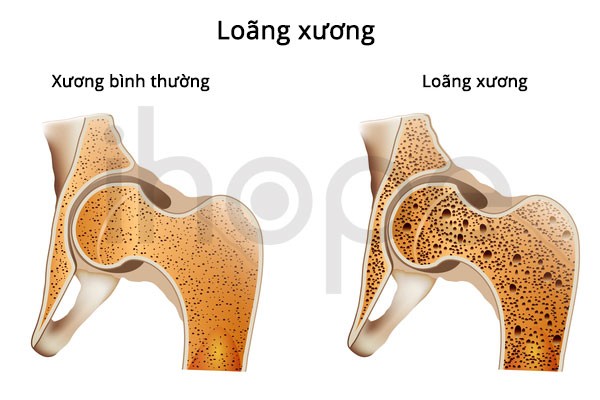
Ảnh: Loãng xương
Nguồn: U.S. National Library of Medicine
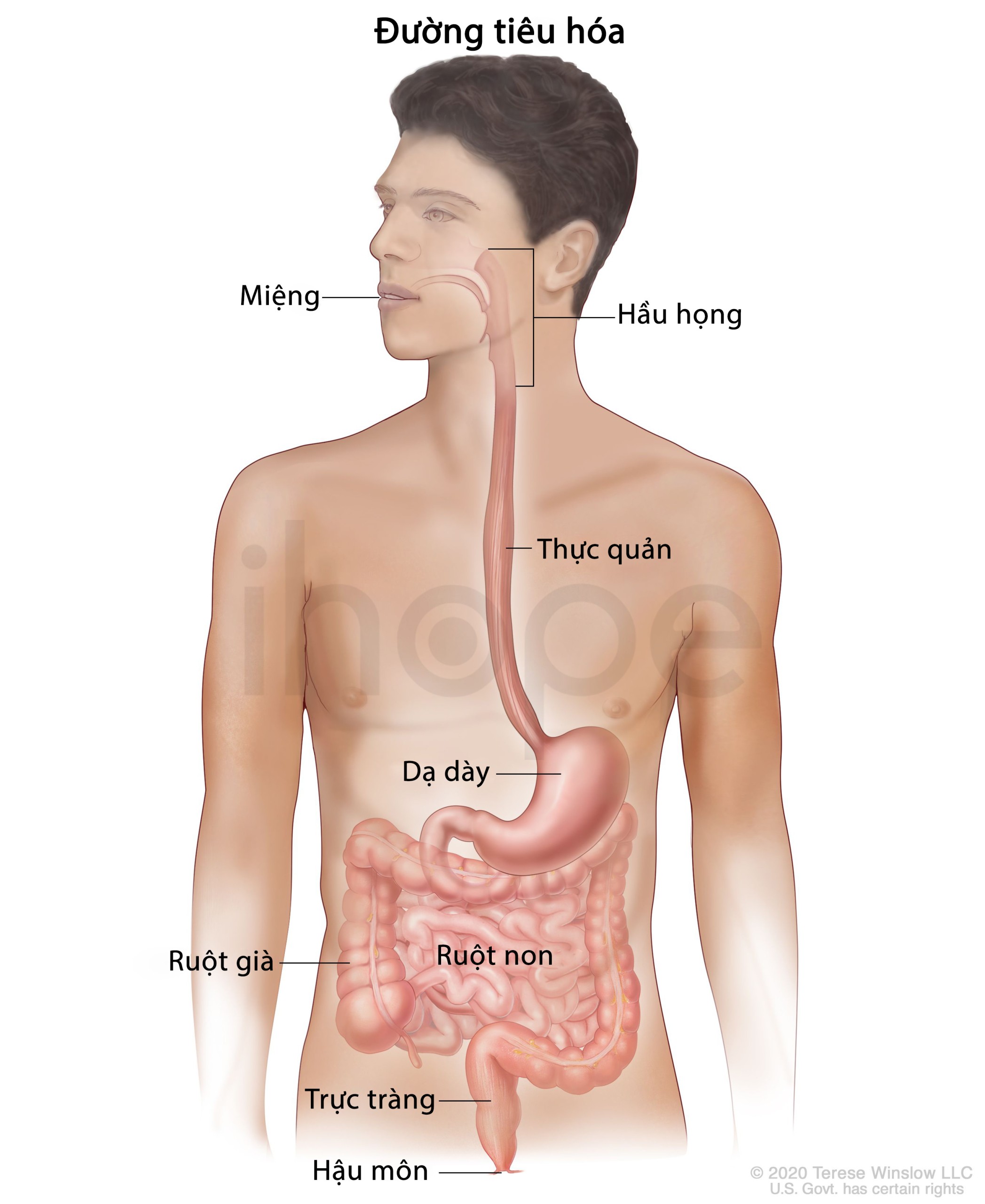
Ảnh: Giải phẫu hệ tiêu hóa
Nguồn: © 2019 Terese Winslow LLC for the National Cancer Institute
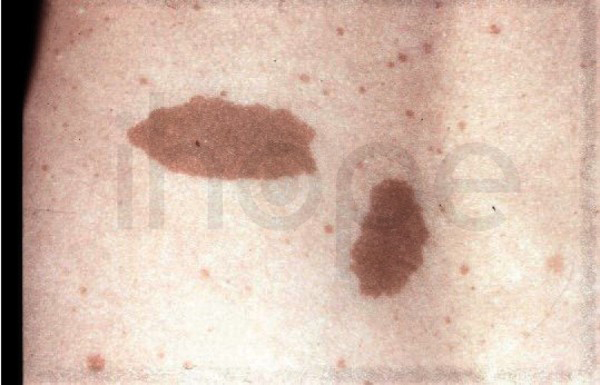
Ảnh: Đốm café-au-lait
Nguồn: GeneReviews, © 1993-2020 University of Washington
Độ phổ biến
Hội chứng Coats cộng rất hiếm gặp. Hiện nay, tỉ lệ mắc bệnh vẫn chưa được thống kê chính xác.
Nguyên nhân
Đột biến gen CTC1 gây ra hội chứng Coats cộng. Gen này cung cấp hướng dẫn tạo ra protein CTC1 có chức năng quan trọng đối với cấu trúc telomere. Telomere là những đoạn ADN ngắn và lặp lại, chúng hiện diện tại các đầu mút của nhiễm sắc thể . Telomere bảo vệ nhiễm sắc thể không bị dính vào nhau hoặc bị đứt gãy, do đó telomere thu ngắn hơn sau mỗi lần phân chia tế bào. Khi telomere ngắn tối đa, tế bào ngừng phân chia hoặc tiến vào quá trình apoptosis.
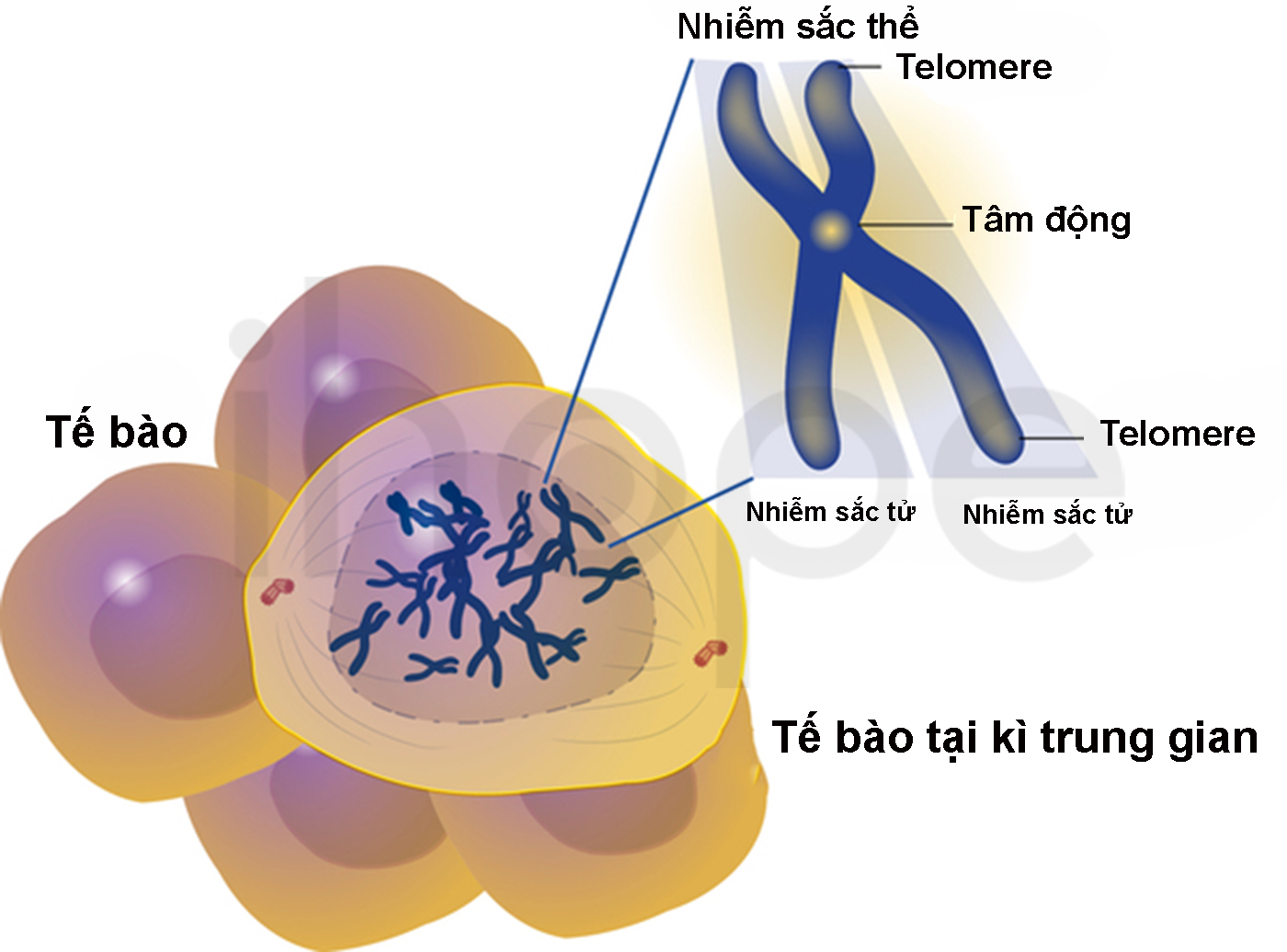
Nguồn: Darryl Leja, NHGRI
Protein CTC1 là tiểu phần của nhóm phức hợp protein CST. Phức hợp này liên quan đến quá trình sao chép telomere, chúng ngăn telomere ngắn đi trong một số tế bào.
Các đột biến gen CTC1 khiến phức hợp CST giảm chức năng, vì vậy quá trình sao chép của telomere bị ảnh hưởng. Tuy nhiên, người ta chưa rõ đột biến gen CTC1 ảnh hưởng đến cấu trúc và chức năng của telomere như thế nào. Một số bệnh nhân mang đột biến gen CTC1 có telomere ngắn bất thường, trong khi những trường hợp khác có telomere không thay đổi kích thước. Người ta đang xác định telomere thay đổi như thế nào trên những người mang đột biến gen CTC1 và vì sao đột biến gây ra các dấu hiệu và triệu chứng khác nhau của hội chứng Coats cộng.
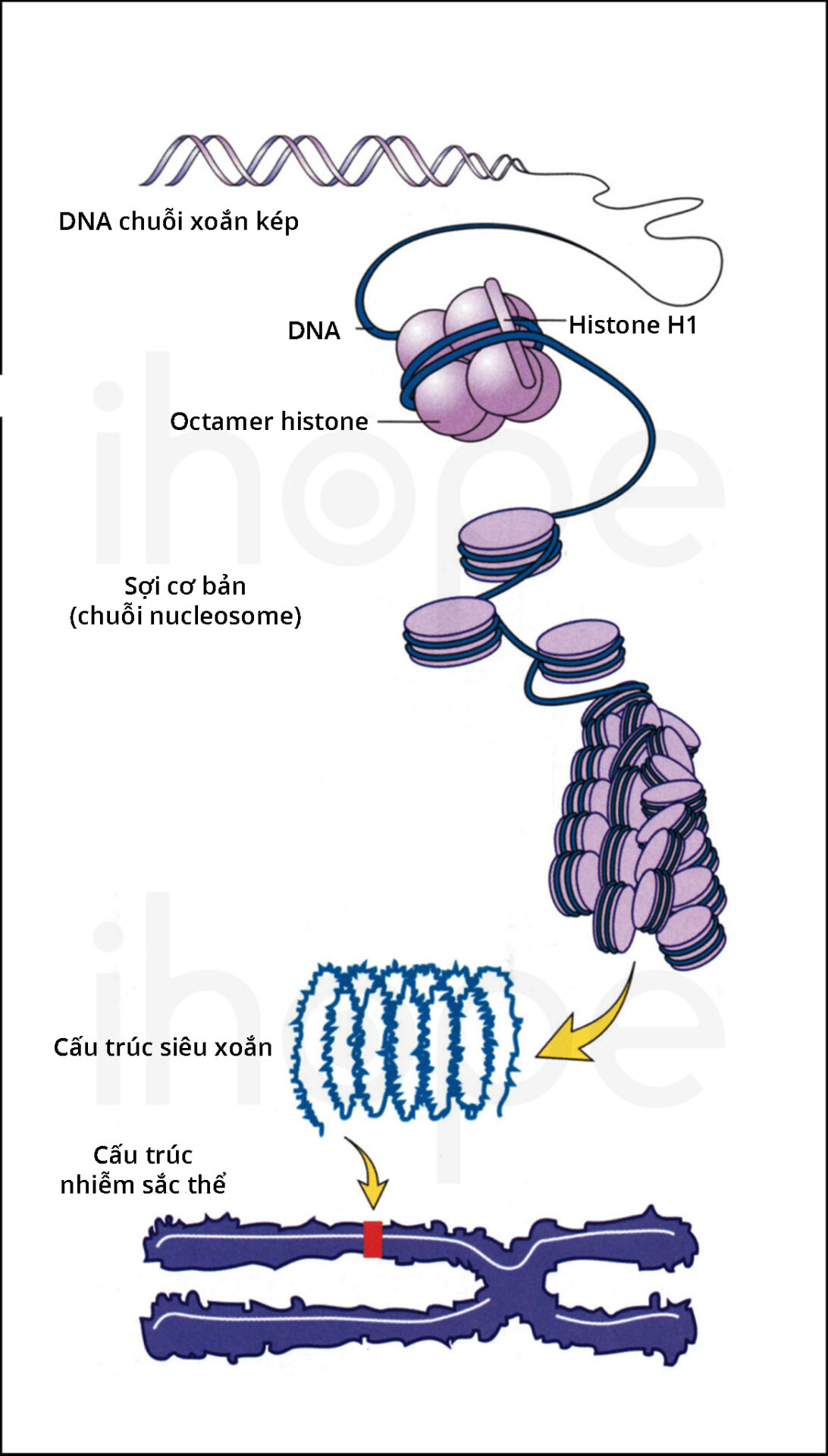
wp-content/uploads/2024/06/Cau-truc-NST.jpg" alt="Cấu Trúc Nst"
Chẩn đoán
Bác sĩ chẩn đoán hột chứng Coats cộng thông qua quan sát các triệu chứng lâm sàng và tiền sử bệnh nhân.
Một số phương pháp chẩn đoán bệnh bao gồm:
- Chụp võng mạc nhằm phát hiện bất thường mạch máu trong võng mạc
- Chụp MRI não cho thấy mức độ biến đổi của chất trắng và kiểm tra sự hiện diện của u nang
- Chụp CT não kết hợp với MRI não nhằm phát hiện vôi hóa và thiếu máu cục bộ
- Xét nghiệm tủy sống và máu có thể phát hiện thiếu máu và giảm tiểu cầu trong một số trường hợp
Ngoài ra, người bệnh có thể thực hiện xét nghiệm di truyền để phát hiện đột biến gen CTCT1, qua đó kết quả chẩn đoán được xác nhận.
Điều trị
Hiện nay chưa có phương pháp điều trị hoàn toàn hội chứng Coats cộng. Các liệu pháp chủ yếu nhằm giảm nhẹ triệu chứng và cải thiện chất lượng đời sống người bệnh. Bác sĩ xây dựng phác đồ điều trị tuỳ thuộc vào mức độ biểu hiện của từng bệnh nhân.
Một số phương pháp điều trị bất thường võng mạc bao gồm:
- Laser quang động vào vùng võng mạc thiếu máu để phá hủy các vùng này, từ đó ngăn ngừa biến chứng.
- Đông lạnh võng mạc dùng nhiệt độ cực lạnh tạo độ kết dính giữa lớp võng mạc bị bong ra và mô bên dưới. Biện pháp này giúp gắn lại võng mạc vào vị trí thích hợp.
Ngoài ra, người bệnh có thể lựa chọn các phương pháp như tiêm nội nhãn thuốc kháng VEGF, tiêm hoặc uống steroid và phẫu thuật mắt.
Đối với bệnh nhân có triệu chứng xuất huyết tiêu hóa đe dọa tính mạng, bác sĩ sẽ tiến hành tiêm tĩnh mạch octreotide. Octreotide có khả năng kiểm soát hiện tượng chảy máu do dị dạng mạch máu.
Dạng di truyền
Hội chứng Coats cộng di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen mang đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh lặn trên nhiễm sắc thể thường sẽ có bố và mẹ mang một bản sao của gen đột biến, nhưng họ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Hội chứng Coats cộng di truyền lặn do đột biến gen CTC1, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. C mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kì.
Các tên gọi khác
- CRMCC
- Cerebroretinal microangiopathy with calcifications and cysts
References
- Genetic Testing Information. Cerebroretinal microangiopathy with calcifications and cysts 1. Retrieved June 24, 2024 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C4552029/
- Genetic and Rare Diseases Information Center. Coats disease. Retrieved June 24, 2024 from https://rarediseases.info.nih.gov/diseases/6121/index
- Catalog of Genes and Diseases from OMIM. CEREBRORETINAL MICROANGIOPATHY WITH CALCIFICATIONS AND CYSTS 1; CRMCC1. Retrieved June 24, 2024 from https://omim.org/entry/612199
- U.S National Library of Medicine. Coats plus syndrome. Retrieved June 24, 2024 from https://medlineplus.gov/genetics/condition/coats-plus-syndrome/
- MalaCards. Cerebroretinal Microangiopathy with Calcifications and Cysts 1 (CRMCC1). Retrieved June 24, 2024 from https://www.malacards.org/card/cerebroretinal_microangiopathy_with_calcifications_and_cysts_1
- National Institute of Health. Coats plus syndrome (cerebroretinal microangiopathy with calcifications and cysts-1): A case report. Retrieved June 24, 2024 from https://pubmed.ncbi.nlm.nih.gov/33010065/
- Orphanet. Coats plus syndrome. Retrieved June 24, 2024 from https://www.orpha.net/en/disease/detail/313838
- Hereditary Ocular Disease. Coats Plus Syndrome. Retrieved June 24, 2024 from https://disorders.eyes.arizona.edu/disorders/coats-plus-syndrome