Teo cơ tủy sống kèm suy hô hấp loại 1 (spinal muscular atrophy with respiratory distress type 1) là bệnh di truyền gây yếu cơ và suy hô hấp. Bệnh thường khởi phát trong giai đoạn sơ sinh và trẻ nhỏ.
Nguồn: © 2006 Terese Winslow LLC for the National Cancer Institute
Biểu hiện lâm sàng
Triệu chứng ban đầu của trẻ bị teo cơ tủy sống kèm suy hô hấp loại 1 gồm:
- Khó thở
- Thở khò khè khi hít vào
- Tiếng khóc yếu
- Ăn uống khó khăn
- Viêm phổi tái phát
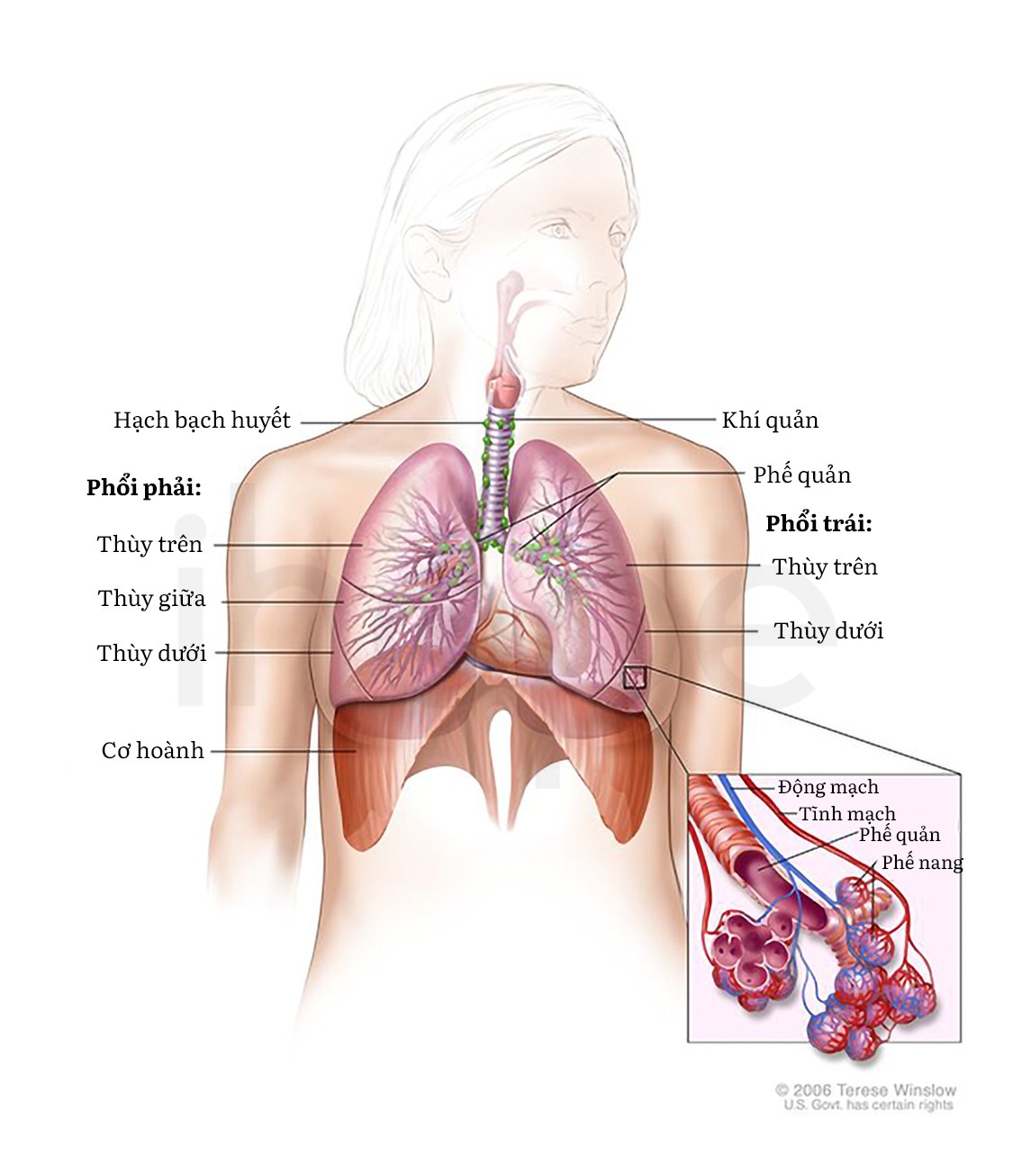
Đối với trẻ sơ sinh mắc bệnh trong độ tuổi từ 6 tuần đến 6 tháng, trẻ có thể đột ngột không thở được do liệt cơ hoành—cơ ngăn cách bụng với khoang ngực. Thông thường, cơ hoành co lại và di chuyển xuống dưới trong quá trình hít vào để phổi nở ra. Trong trường hợp liệt cơ hoành, trẻ cần hỗ trợ thở máy để có thể hô hấp bình thường. Qua 6 tháng tuổi, phần lớn trẻ không biểu hiện triệu chứng liệt cơ hoành.
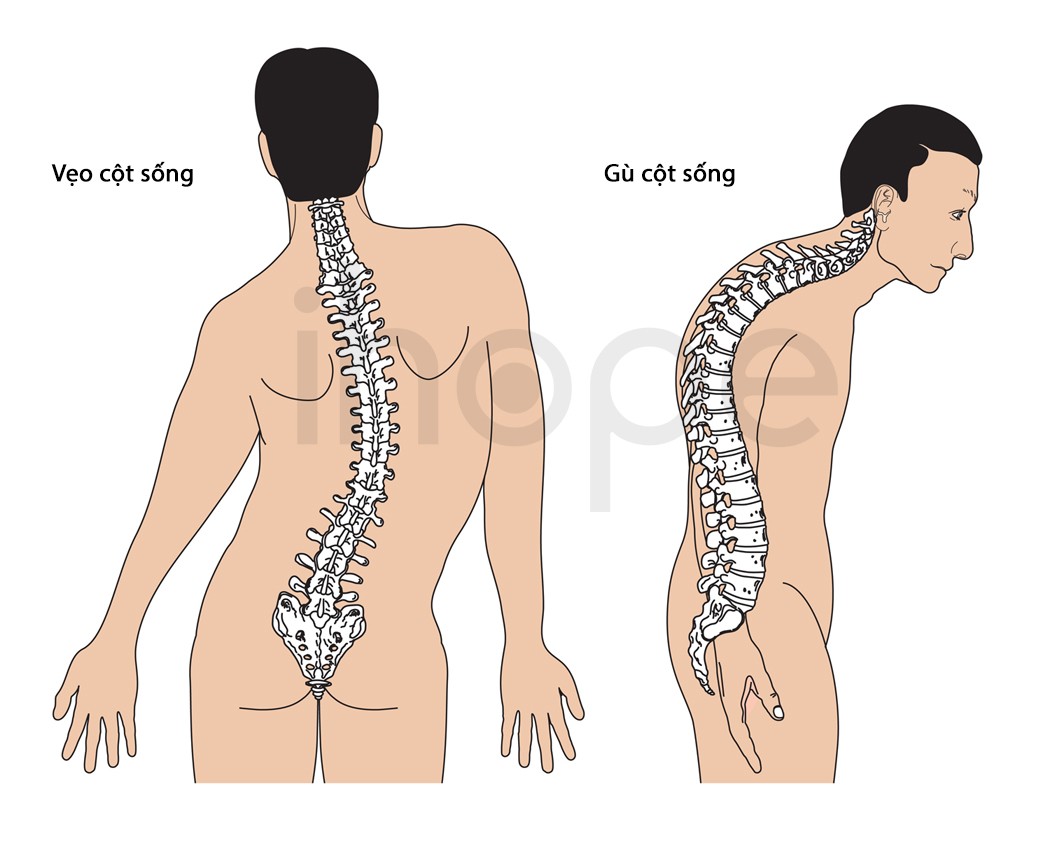
Sau khi bị suy hô hấp, người bệnh có biểu hiện yếu các cơ nằm xa trung tâm cơ thể như cơ tay và cơ chân. Theo thời gian, triệu chứng yếu cơ lan sang tất cả các cơ trên cơ thể. Tuy nhiên, tình trạng yếu cơ ngừng tiến triển sau hai năm. Trong một số trường hợp, người bệnh có thể duy trì chức năng cơ với mức độ thấp, trong khi những trường hợp khác không còn khả năng cử động cơ. Yếu cơ ảnh hưởng nghiêm trọng đến quá trình phát triển vận động của trẻ như ngồi, đứng và đi. Một số trẻ có thể bị cong vẹo cột sống và gù lưng. Sau một tuổi, trẻ mắc bệnh có thể mất phản xạ gân, nghĩa là trẻ không có phản xạ đá về phía trước khi bác sĩ gõ vào đầu gối bằng búa phản xạ.
Nguồn: Blamb/Shutterstock.com
Các triệu chứng khác của teo cơ tủy sống kèm suy hô hấp loại 1 gồm:
- Mất cảm giác đau
- Tăng tiết mồ hôi
- Mất kiểm soát bàng quang và ruột
- Loạn nhịp tim
Độ phổ biến
Teo cơ tủy sống kèm suy hô hấp loại 1 rất hiếm gặp. Hiện nay, chưa có thống kê cụ thể về tỉ lệ mắc bệnh. Hơn 60 trường hợp mắc bệnh đã được ghi nhận trong tài liệu y khoa.
Nguyên nhân
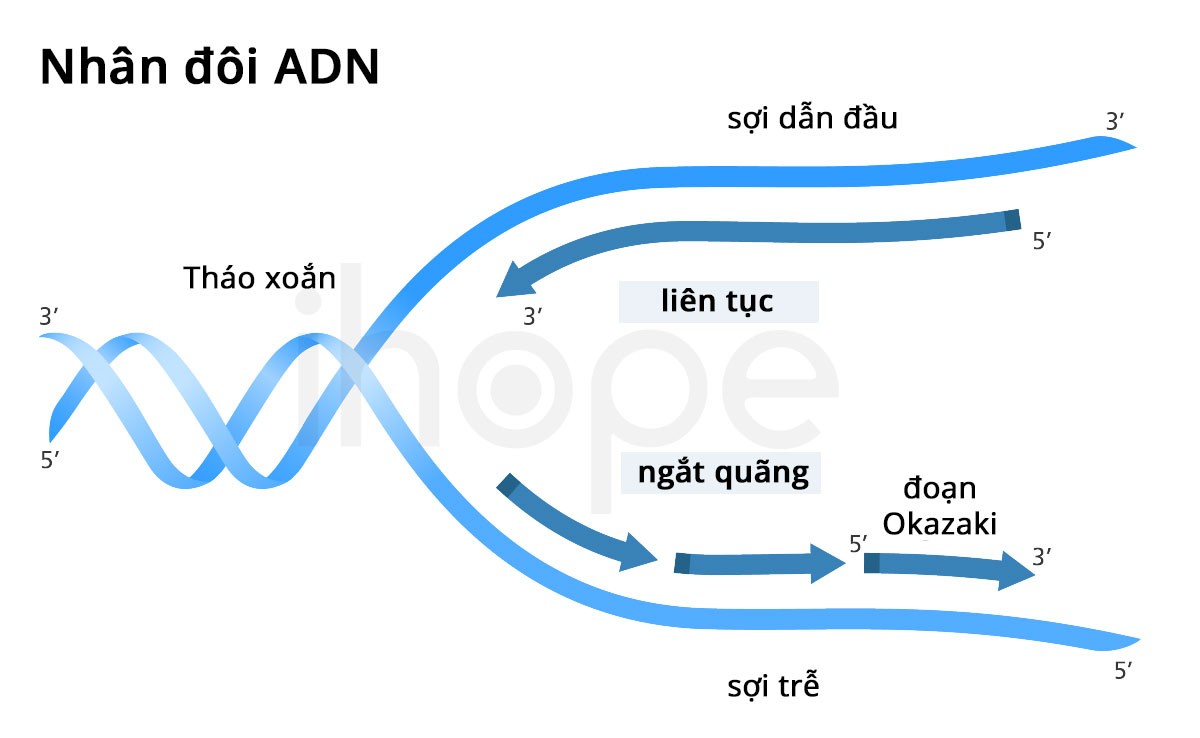
Đột biến gen IGHMBP2 gây ra teo cơ tủy sống kèm suy hô hấp loại 1. Gen này cung cấp hướng dẫn tạo ra protein IGHMBP2 tham gia quá trình sao chép ADN, sản xuất ARN và sản xuất protein.
Nguồn: yourgenome.org
Đột biến gen IGHMBP2 làm giảm khả năng hoạt động của protein tạo thành, do đó chúng ảnh hưởng đến tế bào thần kinh vận động alpha—tế bào chuyên biệt tại thân não và tủy sống. Những tế bào này có chức năng kiểm soát chuyển động của cơ. Protein IGHMBP2 suy giảm chức năng góp phần làm tổn thương các tế bào thần kinh này nên chúng chết dần theo thời gian. Nhiều tế bào thần kinh vận động alpha chết đi tích lũy dần đến mức gây ra các vấn đề hô hấp và suy nhược cơ tiến triển.
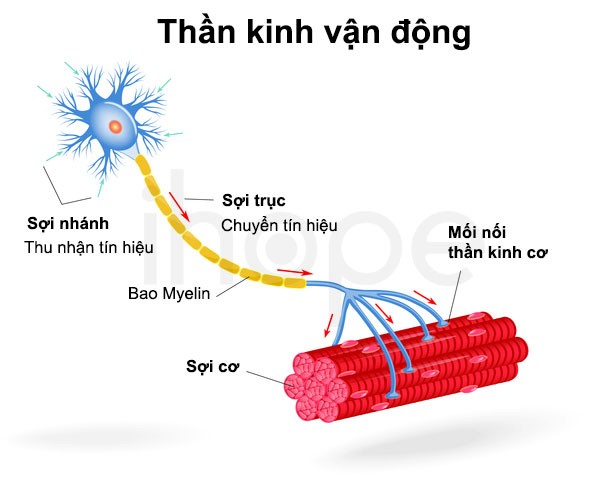
Nguồn: U.S. National Library of Medicine
Nghiên cứu cho thấy lượng protein chức năng do gen IGHMBP2 đột biến tạo ra có thể ảnh hưởng đến mức độ nghiêm trọng của người bệnh. Đối với bệnh nhân còn một phần protein chức năng, họ có xu hướng khởi phát bệnh muộn hơn và duy trì chức năng cơ bắp với mức độ cao hơn.
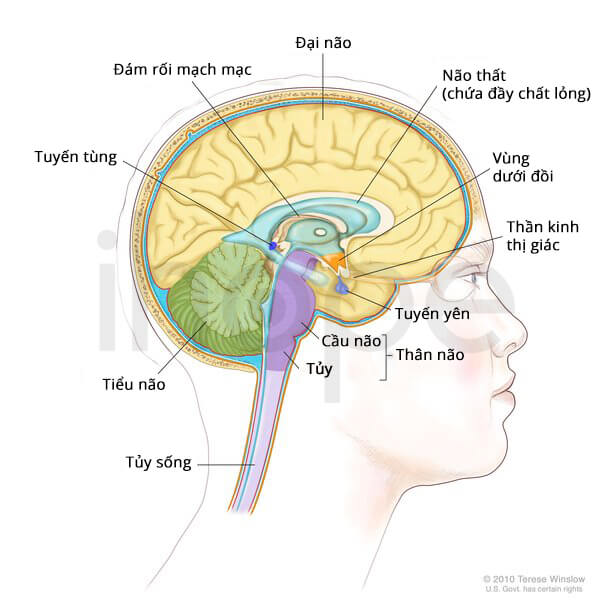
Ảnh: Cấu trúc bên trong của não
Nguồn: Terese Winslow
Chấn đoán
Chẩn đoán teo cơ tủy sống kèm suy hô hấp loại 1 dựa trên biểu hiện lâm sàng kết hợp với xét nghiệm chuyên biệt. Trẻ bị nghi ngờ mắc bệnh nếu suy hô hấp nghiêm trọng và tiến triển nhanh do liệt cơ hoành.
Một số biểu hiện lâm sàng trong chẩn đoán bao gồm:
- Vị trí cơ hoành cao bất thường
- Trẻ sơ sinh bị suy hô hấp
- Gia đình có tiền sử mắc hội chứng đột tử sơ sinh
- Yếu cơ bàn chân và bàn tay
- Co rút khớp xa
Các xét nghiệm chuyên sâu như chụp X-quang, điện cơ đồ (EMG), nghiên cứu dẫn truyền thần kinh (NCS) và sinh thiết cơ có thể được thực hiện để loại trừ nguy cơ mắc những bệnh lí khác. Ngoài ra, người bệnh cần thực hiện xét nghiệm di truyền nhằm phát hiện đột biến gen IGHMBP2, qua đó kết quả chẩn đoán được xác nhận.
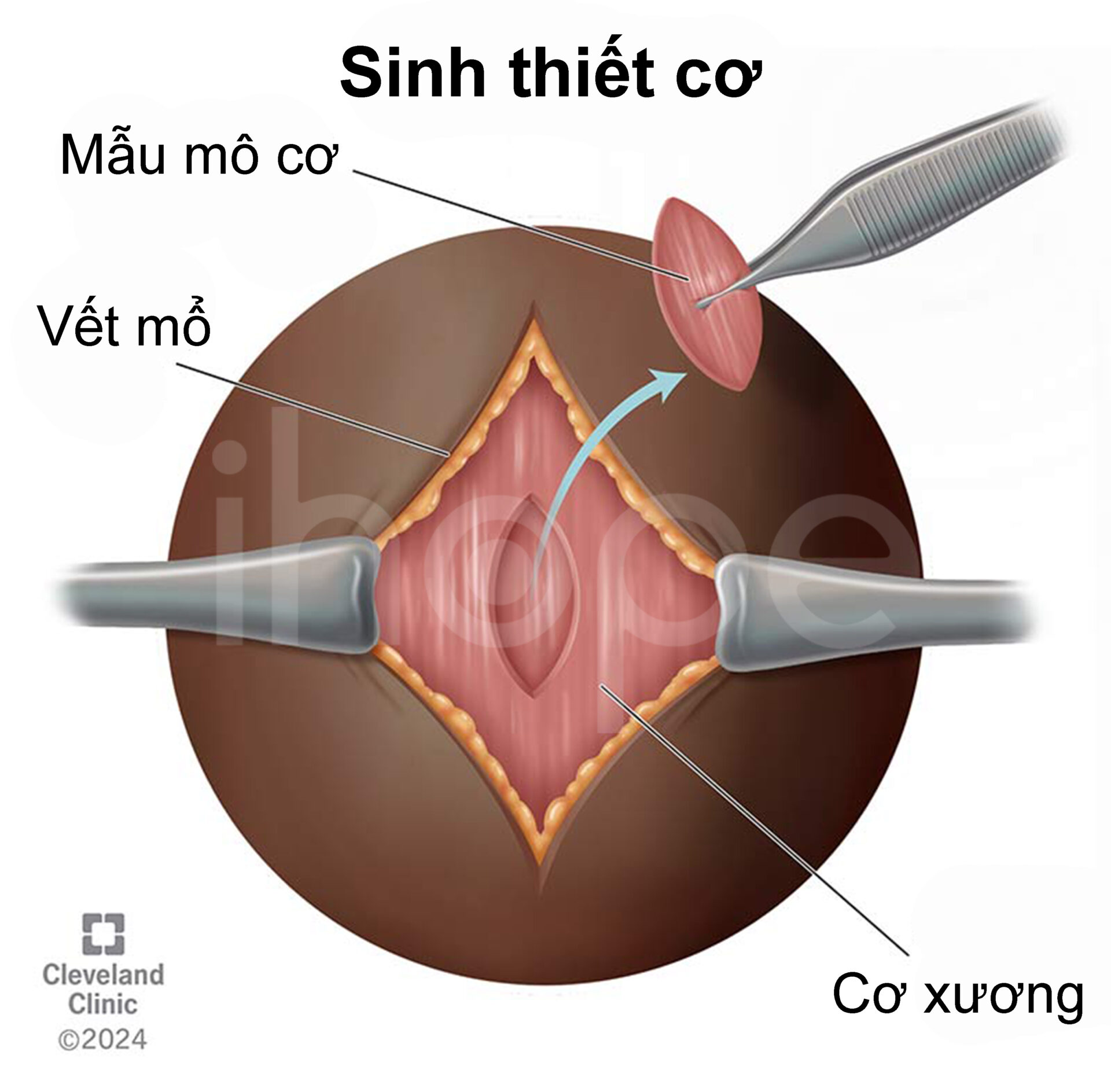
Nguồn: Cleveland Clinic
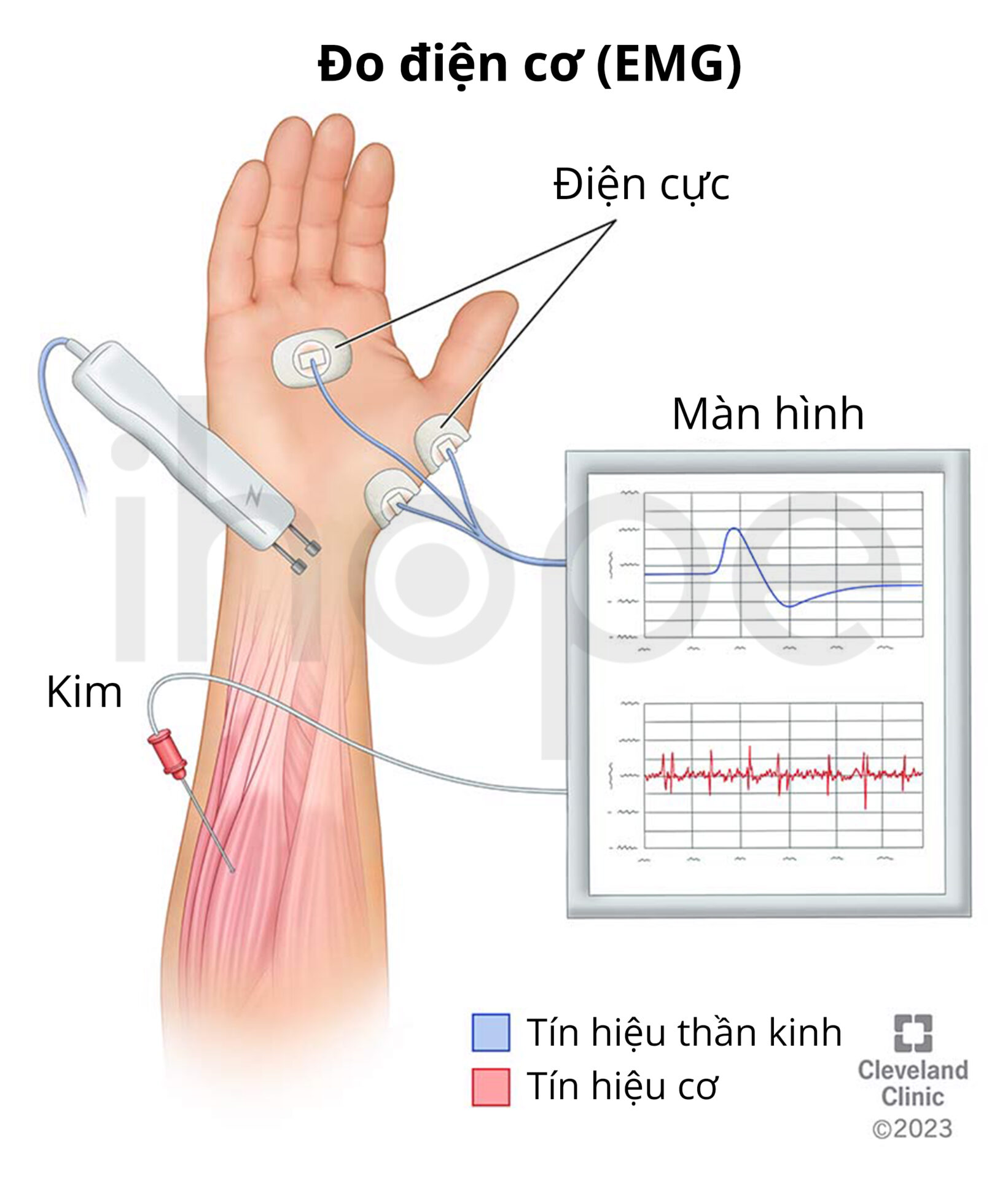
Ảnh: Đo điện cơ
Nguồn: Cleveland Clinic
Điều trị
Hiện nay, chưa có phương pháp điều trị hoàn toàn teo cơ tủy sống kèm suy hô hấp loại 1. Các liệu pháp chủ yếu nhằm giảm nhẹ triệu chứng và cải thiện chất lượng đời sống người bệnh. Bệnh nhân liệt cơ hoành cần được hỗ trợ thở máy sớm. Nếu không được thở máy, phần lớn người bệnh sẽ tử vong do suy hô hấp trước 13 tháng tuổi. Nhiễm trùng đường hô hấp tái phát được điều trị bằng liệu pháp kháng sinh và các biện pháp phòng ngừa dự phòng khác.
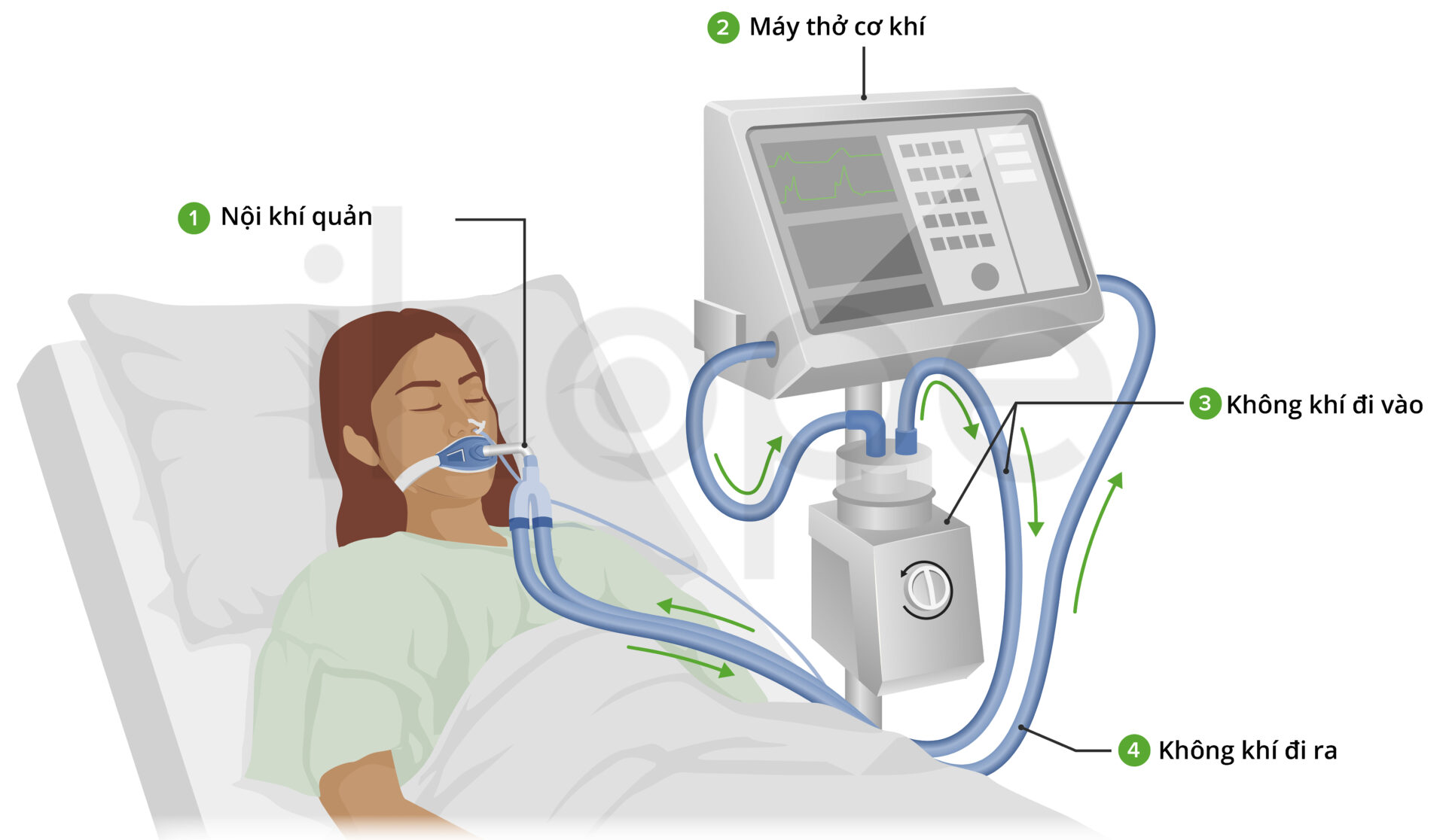
Nguồn: SafarMuslim
Ngoài ra, người bệnh có biểu hiện khó nuốt và tiêu hóa thức ăn do yếu cơ và rối loạn chức năng đường tiêu hóa. Do đó, bác sĩ có thể đặt ống thông mũi dạ dày để cung cấp chất dinh dưỡng. Ngoài ra, người bệnh cần tiến hành vật lí trị liệu nhằm duy trì sức mạnh cơ bắp.
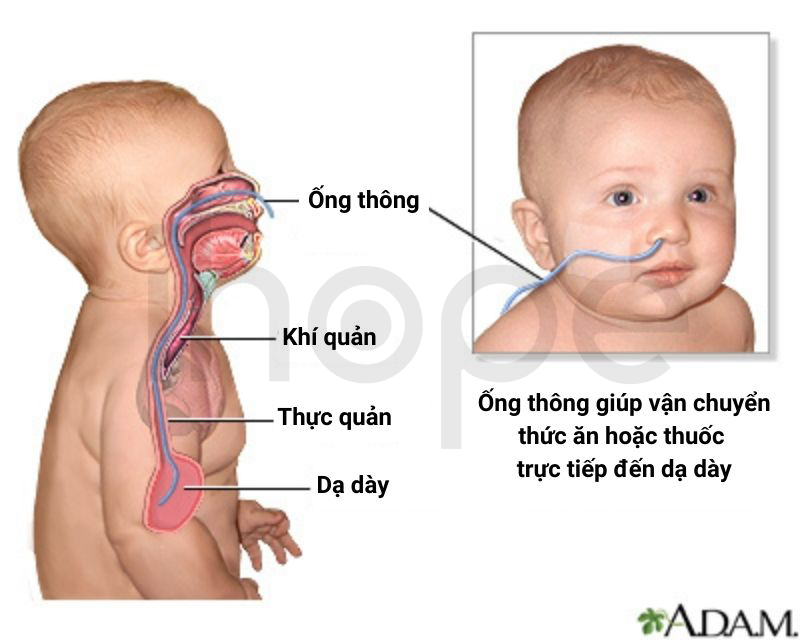
Nguồn: ADAM
Dạng di truyền
Teo cơ tủy sống kèm suy hô hấp loại 1 di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh di truyền gen lặn trên nhiễm sắc thể thường có bố và mẹ mang một bản sao của gen đột biến, nhưng họ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Teo cơ tủy sống kèm suy hô hấp loại 1 di truyền lặn đột biến gen IGHMBP2. Cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kì.
Các tên gọi khác
- Autosomal recessive distal spinal muscular atrophy 1
- DHMN6
- Diaphragmatic spinal muscular atrophy
- Distal hereditary motor neuronopathy type VI
- Distal spinal muscular atrophy type 1
- DSMA1
- HMN6
- HMNVI
- Severe infantile axonal neuropathy with respiratory failure
- SIANRF
- SMARD1
- Spinal muscular atrophy with respiratory distress
References
- Genetic Testing Information. Autosomal recessive distal spinal muscular atrophy 1. Retrieved March 03, 2025 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C1858517/
- Genetic and Rare Diseases Information Center. Autosomal recessive distal spinal muscular atrophy 1. Retrieved March 03, 2025 from https://rarediseases.info.nih.gov/diseases/8592/index
- Catalog of Genes and Diseases from OMIM. NEURONOPATHY, DISTAL HEREDITARY MOTOR, AUTOSOMAL RECESSIVE 1; HMNR1. Retrieved March 03, 2025 from https://omim.org/entry/604320
- U.S National Library of Medicine. Spinal muscular atrophy with respiratory distress type 1. Retrieved March 03, 2025 from https://medlineplus.gov/genetics/condition/spinal-muscular-atrophy-with-respiratory-distress-type-1/
- National Institute of Health. Spinal muscular atrophy with respiratory distress type 1: Clinical phenotypes, molecular pathogenesis and therapeutic insights. Retrieved March 03, 2025 from https://pubmed.ncbi.nlm.nih.gov/31802621/
- National Institute of Neurological Disorders and Stroke. Spinal Muscular Atrophy with Respiratory Distress Type 1. Retrieved March 03, 2025 from https://rarediseases.org/rare-diseases/spinal-muscular-atrophy-with-respiratory-distress/
- Orphanet. Spinal muscular atrophy with respiratory distress type 1. Retrieved March 03, 2025 from https://www.orpha.net/en/disease/detail/98920
- National Institute of Health. Current understanding of and emerging treatment options for spinal muscular atrophy with respiratory distress type 1 (SMARD1). Retrieved March 03, 2025 from https://pubmed.ncbi.nlm.nih.gov/32123965/