
Aldosterone là hormone có chức năng chuyển hóa muối (mineralocorticoid), nó thuộc nhóm nội tiết tố steroid được tạo ra từ vỏ tuyến thượng thận (corticosteroid). Aldosterone giúp cơ thể tái hấp thụ natri và nước đồng thời bài tiết kali qua thận, da, ruột và nước tiểu. Do đó, cơ thể tăng khả năng giữ nước và dịch ngoại bào. Ngoài ra, huyết áp có thể được điều hòa bằng cách thay đổi nồng độ aldosterone.
Giả giảm aldosterone loại 1 (pseudohypoaldosteronism type 1) là hội chứng di truyền hiếm gặp khiến ống thận và một số mô khác không đáp ứng với aldosterone. Do đó, kali tích tụ trong cơ thể nhưng natri và nước lại được bài tiết quá mức dẫn đến tình trạng hạ huyết áp. Hội chứng giả giảm aldosterone khởi phát các biểu hiện của chứng giảm hormone aldosterone. Tuy nhiên, thực tế bệnh nhân luôn có nồng độ cao hormone aldosterone.
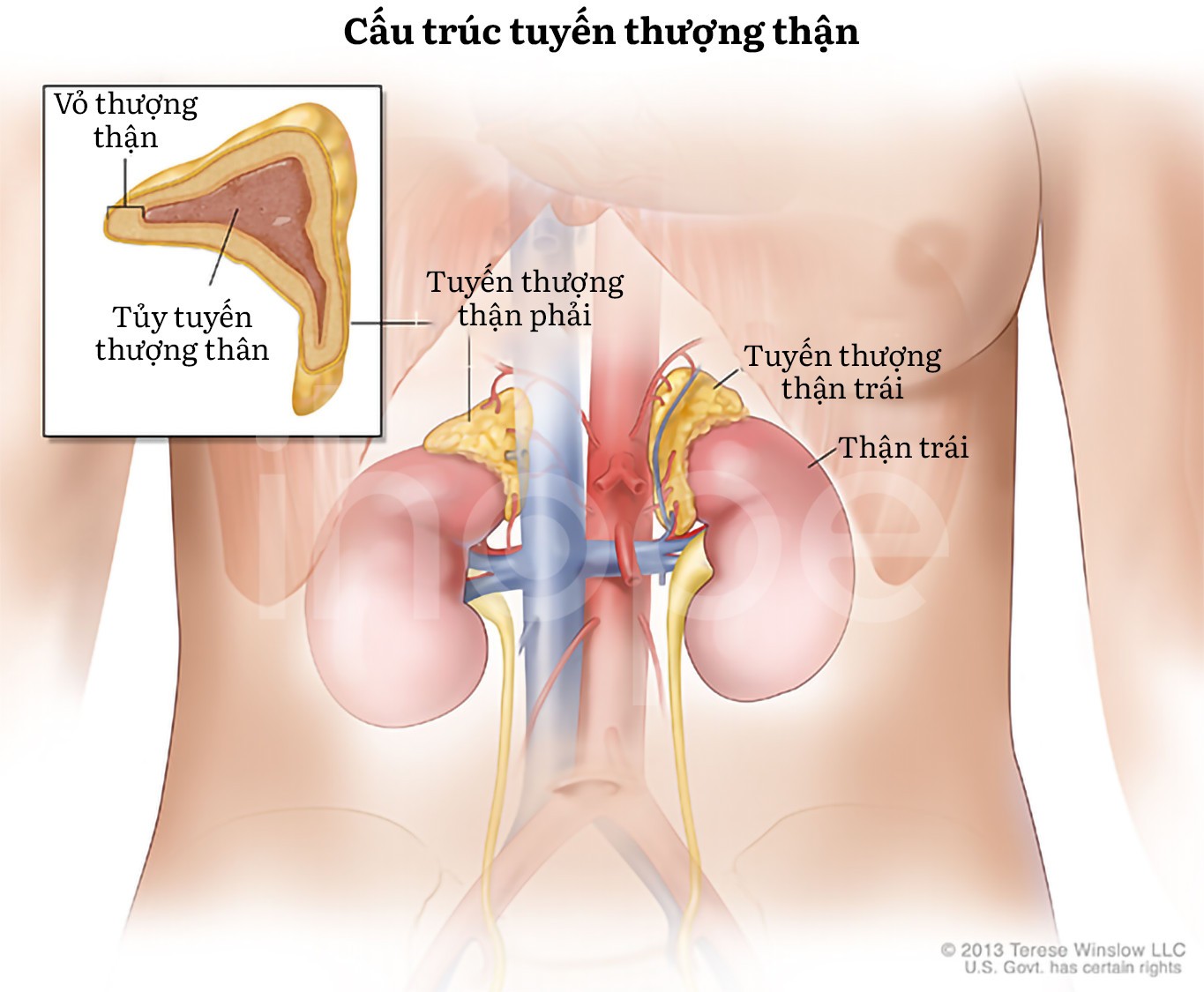
Nguồn: U.S. National Library of Medicine
Biểu hiện lâm sàng
Hội chứng giả giảm aldosterone được phân loại dựa trên mức độ nghiêm trọng, các đột biến gen liên quan và cơ chế di truyền.
Giả giảm aldosterone loại 1 dạng di truyền theo kiểu trội trên nhiễm sắc thể thường (giả giảm aldosteron loại 1 liên quan thận) chỉ gây mất natri thận. Dạng bệnh này tương đối nhẹ và thường cải thiện trong giai đoạn trẻ nhỏ. Bệnh có thể tự thuyên giảm theo thời gian.
Các biểu hiện của hội chứng giả giảm aldosterone loại 1 liên quan thận bao gồm:
- Hạ natri máu
- Tăng kali máu
- Nồng độ axit cao trong máu (nhiễm toan chuyển hóa)
- Nồng độ renin và aldosterone trong huyết tương tăng cao
Giả giảm aldosterone loại 1 dạng di truyền theo kiểu lặn trên nhiễm sắc thể thường (giả giảm aldosteron loại 1 tổng quát) gây mất natri thận và nhiều cơ quan khác bao gồm phổi, thận, ruột kết, tuyến mồ hôi và tuyến nước bọt. Bệnh dạng này có các triệu chứng nghiêm trọng và kéo dài đến tuổi trưởng thành. Những biểu hiện không thuyên giảm theo thời gian.
Một số triệu chứng của hội chứng giả giảm aldosterone loại 1 tổng quát bao gồm:
- Các biểu hiện của dạng liên quan thận
- Bài tiết natri qua mồ hôi, nước tiểu và phân cao bất thường
- Chảy nước mũi dai dẳng, ho
- Nhiễm trùng đường thở, suy hô hấp
- Thở nhanh và khò khè
- Tổn thương da (do viêm cấu trúc eccrine)
Những biểu hiện khởi phát từ giai đoạn sơ sinh do cả hai dạng bệnh gây ra:
- Mất muối và nước
- Không tăng cân, chậm lớn
- Mệt mỏi, nôn mửa
- Nhiễm trùng đường hô hấp dưới
- Loạn nhịp tim
- Nhiễm trùng phổi tái phát
- Tổn thương da
Độ phổ biến
Giả giảm aldosterone loại 1 liên quan thận có độ phổ biến cao hơn. Người ta ước tính tỷ lệ mắc hội chứng dạng này khoảng 1/66.000 trẻ sơ sinh. Đối với giả giảm aldosterone loại 1 tổng quát, tỷ lệ mắc khoảng 1/166.000 trẻ sơ sinh tại Anh.
Nguyên nhân
Protein thụ thể mineralocorticoid có khả năng điều chỉnh các protein chuyên biệt trên màng tế bào nhằm kiểm soát quá trình vận chuyển natri hoặc kali. Thụ thể này làm tăng số lượng và hoạt tính các protein trên màng tế bào để đáp ứng với nồng độ natri thấp khi có sự hiện diện của hormone aldosterone. Thụ thể mineralocorticoid kiểm soát một số kênh trên màng tế bào thận bao gồm kênh ENaC và kênh đồng vận chuyển kali–natri. Những kênh này duy trì nồng độ natri ổn định thông quá trình tái hấp thu và loại bỏ kali khỏi cơ thể.
Đột biến gen NR3C2 gây ra hội chứng giả giảm aldosterone loại 1 dạng di truyền trội trên nhiễm sắc thể thường. Gen NR3C2 cung cấp hướng dẫn tạo ra protein thụ thể mineralocorticoid. Đột biến này khiến thụ thể mineralocorticoid giảm hoạt động hoặc mất chức năng. Do nó, quá trình điều chỉnh hoạt động các protein chuyên biệt trên màng tế bào bị gián đoạn, từ đó gây ra biểu hiện hạ natri và tăng kali máu.
Đột biến gen SCNN1A, SCNN1B và SCNN1G gây ra hội chứng giả giảm aldosterone loại 1 di truyền lặn trên nhiễm sắc thể thường. Những gen này cung cấp hướng dẫn tạo ra các tiểu đơn vị của phức hợp kênh natri biểu mô. Kênh này vận chuyển natri vào trong tế bào. Đột biến gen SCNN1A, SCNN1B và SCNN1G khiến các kênh natri biểu mô mất chức năng. Tình trạng hạ natri và tăng kali khởi phát các triệu chứng của hội chứng giả giảm aldosterone loại 1 tổng quát như nhiễm trùng phổi và tổn thương da.
Chẩn đoán
Hội chứng giả giảm aldosterone loại 1 được chẩn đoán dựa trên các xét nghiệm sinh hóa lâm sàng bao gồm:
- Xét nghiệm thể tích tuần hoàn
- Đo hàm lượng natri và kali máu
- Khảo sát nồng độ renin và aldosterone
Xét nghiệm di truyền nhằm xác định các đột biến gen liên quan giúp xác nhận chẩn đoán. Xét nghiệm di truyền sơ sinh từ máu cuống rốn có thể cho phép phát hiện sớm trẻ sơ sinh mắc bệnh do gia đình có đột biến liên quan đến hội chứng.
Điều trị
Hiện nay chưa có phương pháp điều trị hoàn toàn hội chứng giả giảm aldosterone loại 1. Các liệu pháp giúp giảm nhẹ các triệu chứng và cải thiện chất lượng đời sống bệnh nhân.
Một số phương pháp điều trị có thể bao gồm:
- Liệu pháp dinh dưỡng: tuân thủ chế độ ăn nhiều natri giúp duy trì thể tích tuần hoàn và tăng bài tiết kali máu.
- Sử dụng thuốc: nếu chế độ ăn kiêng không hiệu quả, bác sĩ có thể chỉ định sử dụng fludrocortisone 0,5 đến 1,0 mg uống hai lần/ngày hoặc 1 đến 2 mg uống một lần/ngày.
Dạng di truyền
Hội chứng giả giảm aldosterone loại 1 liên quan thận xảy ra do đột biến gen NR3C2 di truyền theo kiểu trội trên nhiễm sắc thể thường. Do đó, một bản sao của gen trong mỗi tế bào đủ để gây ra bệnh. Phần lớn người bệnh thừa hưởng đột biến từ cha hoặc mẹ bị ảnh hưởng.

Nguồn: U.S. National Library of Medicine
Hội chứng giả giảm aldosterone loại 1 tổng quát do đột biến gen SCNN1A , SCNN1B và SCNN1G di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh lặn trên nhiễm sắc thể thường sẽ có bố và mẹ mang một bản sao của gen đột biến, nhưng bố mẹ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Trường hợp bệnh di truyền trội trên nhiễm sắc thể thường, nếu cha hoặc mẹ mắc bệnh, con sinh ra sẽ có 50% khả năng di truyền bệnh. Do đó, để bảo đảm 100% khả năng con không bị bệnh, cha mẹ có thể chọn phương pháp thụ tinh nhân tạo IVF và sàng lọc phôi PGS/PGD. Các thành viên trong gia đình nên khám sức khỏe và tầm soát bệnh định kỳ nếu có thành viên mắc bệnh. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh.
Bệnh di truyền lặn, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kỳ.
Các tên gọi khác
- PHA1
References
- Genetic Testing Information. Autosomal dominant pseudohypoaldosteronism type 1. Retrieved 30 June 2023 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C1449842/?_ga=2.183271398.1310707250.1688098504-41285715.1684139855
- Genetic and Rare Diseases Information Center. Renal pseudohypoaldosteronism type 1. Retrieved 30 June 2023 from https://rarediseases.info.nih.gov/diseases/9145/autosomal-dominant-pseudohypoaldosteronism-type-1
- Catalog of Genes and Diseases from OMIM. PSEUDOHYPOALDOSTERONISM, TYPE I, AUTOSOMAL DOMINANT; PHA1A. Retrieved 30 June 2023 from https://omim.org/entry/177735
- U.S National Library of Medicine. Pseudohypoaldosteronism type 1. Retrieved 30 June 2023 from https://medlineplus.gov/genetics/condition/pseudohypoaldosteronism-type-1/
- Frontiers. Case Report: Newborns With Pseudohypoaldosteronism Secondary to Excessive Gastrointestinal Losses Through High Output Stoma. Retrieved 30 June 2023 from https://www.frontiersin.org/articles/10.3389/fped.2021.773246/full
- MalaCards. Pseudohypoaldosteronism Type 1 (PHA1). Retrieved 30 June 2023 from https://www.malacards.org/card/pseudohypoaldosteronism_type_1
- MSD Manuals. Pseudohypoaldosteronism Type I. Retrieved 30 June 2023 from https://www.msdmanuals.com/professional/genitourinary-disorders/renal-transport-abnormalities/pseudohypoaldosteronism-type-i
- National Organization for Rare Disorders. Pseudohypoaldosteronism Foundation. Retrieved 30 June 2023 from https://rarediseases.org/non-member-patient/pseudohypoaldosteronism-foundation/
- Orphanet. Pseudohypoaldosteronism type 1. Retrieved 30 June 2023 from https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=756



