
Hội chứng Gordon Holmes (Gordon Holmes syndrome) là bệnh di truyền hiếm gặp. Bệnh gây ra các vấn đề liên quan đến sinh sản và thần kinh.
Biểu hiện lâm sàng
Một trong những biểu hiện đặc trưng của hội chứng Gordon Holmes là suy sinh dục do giảm gonadotropin. Phần lớn người bệnh có triệu chứng dậy thì chậm.
Các dấu hiệu điển hình của tuổi dậy thì bao gồm:
- Nam giới: mọc lông mặt, giọng nói trầm hơn
- Nữ giới: có chu kì kinh nguyệt, ngực phát triển
Trong một số ít trường hợp, người bệnh không bao giờ trải qua giai đoạn dậy thì. Đôi khi, những bệnh nhân dậy thì bình thường lại xuất hiện các triệu chứng khác liên quan đến sinh sản.
Vào đầu giai đoạn trưởng thành, người bệnh khởi phát các biểu hiện về thần kinh bao gồm nói chuyện khó khăn. Khi bệnh tiến triển nghiêm trọng, bệnh nhân sẽ khó giữ thăng bằng, khó phối hợp các động tác dẫn đến các bất lợi trong hoạt động sinh hoạt hằng ngày và cần sử dụng các dụng cụ hỗ trợ như xe lăn. Trong một số trường hợp, người bệnh xuất hiện các dấu hiệu liên quan đến trí nhớ và giảm chức năng trí tuệ.
Độ phổ biến
Hội chứng Gordon Holmes rất hiếm gặp nên tỉ lệ mắc bệnh chưa được xác định.
Nguyên nhân
Đột biến gen RNF216 hoặc PNPLA6 gây ra hội chứng Gordon Holmes. Trong một số trường hợp, người bệnh không mang đột biến tại các gen này. Do đó, người ta cho rằng đột biến trên các gen khác cũng có thể gây bệnh.
Gen RNF216 hướng dẫn tạo ra protein tham gia vào quá trình ubiquitin hóa. Trong quá trình này, các protein không cần thiết được đánh dấu bằng phân tử ubiquitin. Sau đó, ubiquitin báo hiệu để cơ thể phân hủy protein này. Trong số các protein được đánh dấu bởi RNF216, một protein hiện diện trong tế bào thần kinh. Nó có vai trò quan trọng đối với tính dẻo của khớp thần kinh—khả năng thay đổi và thích nghi theo thời gian của các khe synapse để đáp ứng với kích thích. Quá trình này rất cần thiết cho khả năng học tập và ghi nhớ.
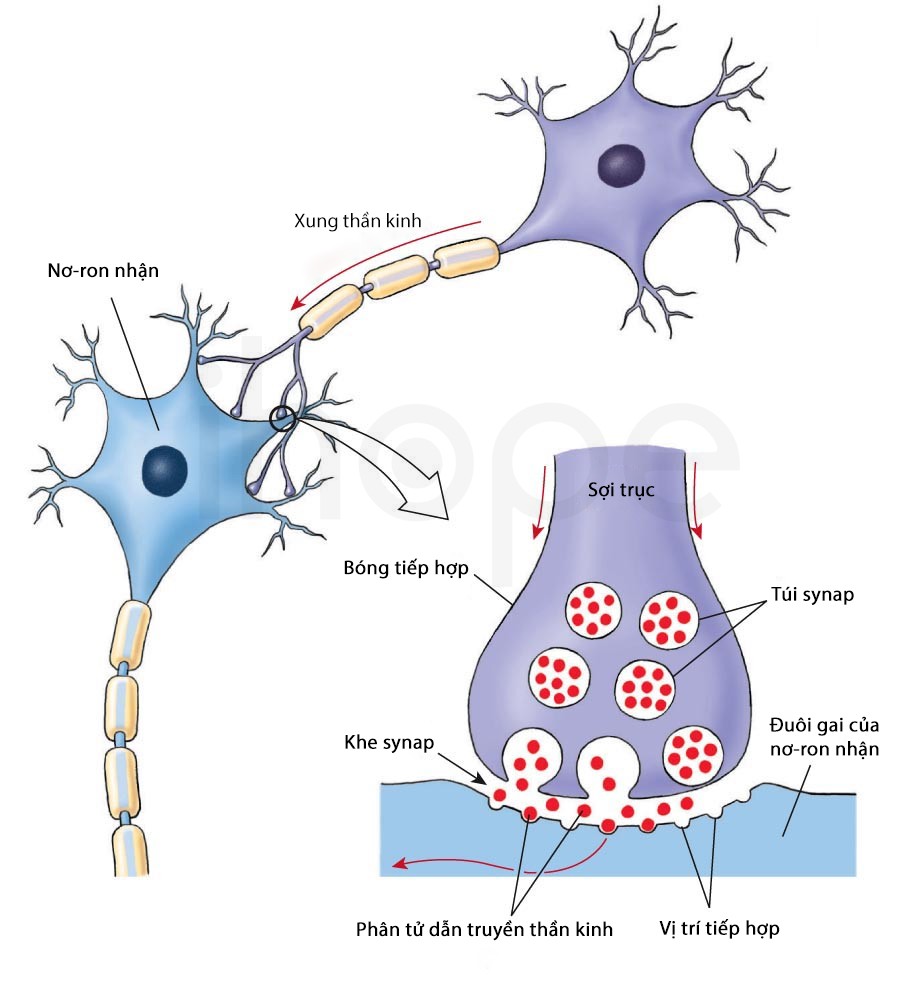
Nguồn: socratic.org
Đột biến gen RNF216 làm giảm khả năng gắn thẻ các protein không cần thiết của protein RNF216. Quá trình phân hủy protein thần kinh giảm gây gián đoạn các kết nối tại khe synapse. Hiện tượng này góp phần gây ra triệu chứng mất trí của người mắc hội chứng Gordon Holmes. Cơ chế đột biến gen RNF216 gây ra triệu chứng suy sinh dục do giảm gonadotropin và mất điều hòa tiểu não chưa được xác định.
Gen PNPLA6 cung cấp hướng dẫn tạo ra protein NTE (neuropathy target esterase) có chức năng điều chỉnh lượng lipid tạo nên màng ngoài tế bào. Nồng độ lipid này sẽ quyết định tính ổn định và chức năng của màng tế bào. Protein NTE hiện diện chủ yếu trong hệ thần kinh. Nó duy trì tính ổn định của màng bao quanh tế bào thần kinh. Ngoài ra, NTE cũng tham gia giải phóng hormone từ tuyến yên. Quá trình này đòi hỏi những thay đổi cụ thể tại màng tế bào. Tuyến yên nằm trong gốc não, nó sản xuất ra một số hormone bao gồm hormone có chức năng điều phối quá trình phát triển và tăng trưởng sinh dục.
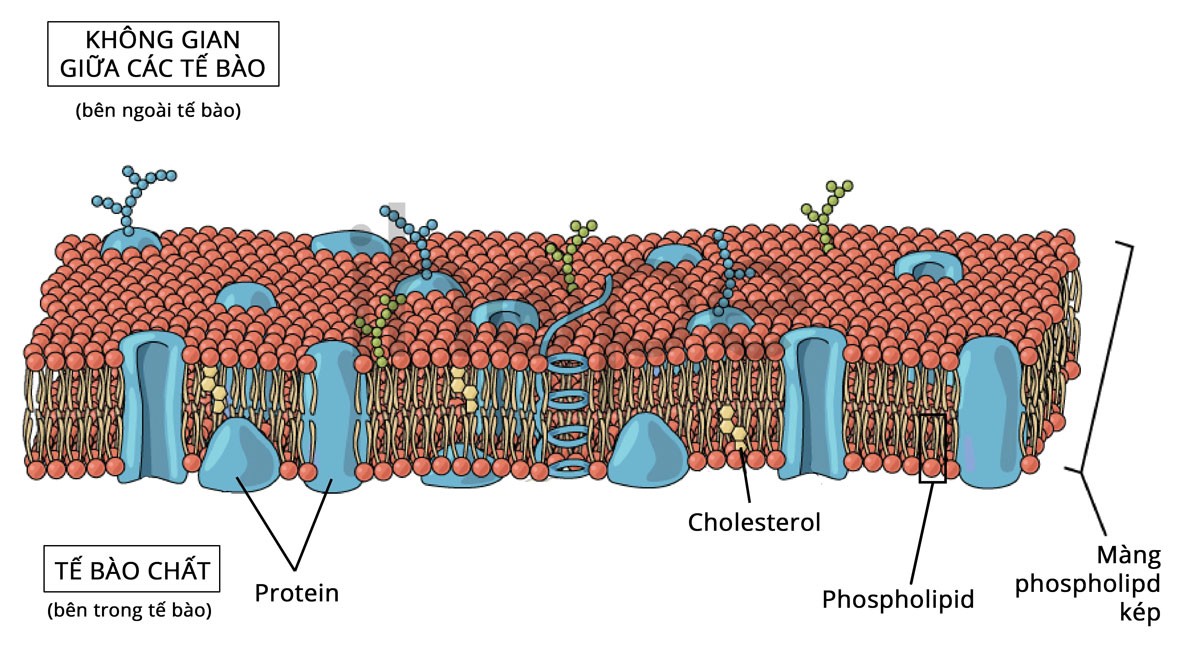
Nguồn: OpenStax Biology
Đột biến gen PNPLA6 khiến chức năng của protein NTE giảm đi. Từ đó, nồng độ lipid trong màng tế bào thay đổi làm tổn thương các tế bào thần kinh trong não. Vì vậy, người bệnh có các biểu hiện như mất điều hòa tiểu não, suy sinh dục do giảm gonadotropin. Người bệnh có đột biến gen PNPLA6 thường không có biểu hiện mất trí.
Chẩn đoán
Bác sĩ nghi ngờ bệnh nhân mắc hội chứng Gordon Holmes thông qua biểu hiện kết hợp hiếm gặp của triệu chứng mất điều hòa và suy sinh dục do giảm gonadotropin. Đồng thời, bác sĩ cũng kiểm tra các dấu hiệu điển hình của giai đoạn dậy thì.
Để kết quả chẩn đoán chính xác hơn, bệnh nhân cần thực hiện một số xét nghiệm như:
- Xét nghiệm nội tiết: nhằm kiểm tra nồng độ hormone kích thích nang trứng, hormone hoàng thể, testosterone, estrogen và xác định triệu chứng suy sinh dục do giảm gonadotropin
- Chụp cộng hưởng từ não: nhằm phát hiện dấu hiệu teo tiểu não, teo vỏ não hoặc giảm mật độ chất trắng
Khi cần thiết, người bệnh có thể thực hiện xét nghiệm di truyền để xác nhận chẩn đoán.
Điều trị
Hiện nay, chưa có phương pháp điều trị hoàn toàn hội chứng Gordon Holmes. Các liệu pháp chủ yếu làm giảm triệu chứng và cải thiện chất lượng cuộc sống của người bệnh.
Dạng di truyền
Hội chứng Gordon Holmes di truyền theo kiểu lặn trên nhiễm sắc thể thường, nghĩa là cả hai bản sao của gen trong mỗi tế bào đều có đột biến. Cha mẹ của một cá nhân mắc bệnh lặn trên nhiễm sắc thể thường đều mang một bản sao của gen đột biến, nhưng họ thường không biểu hiện dấu hiệu và triệu chứng của bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Hội chứng Gordon Holmes di truyền lặn đột biến gen RNF216 hoặc PNPLA6, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kì.
Các tên gọi khác
- Cerebellar ataxia and hypogonadotropic hypogonadism
- Deficiency of luteinizing hormone-releasing hormone with ataxia
- LHRH deficiency and ataxia
References
- Genetic Testing Information. Cerebellar ataxia-hypogonadism syndrome. Retrieved December 13, 2024 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C1859305/
- Genetic and Rare Diseases Information Center. Cerebellar ataxia-hypogonadism syndrome. Retrieved December 13, 2024 from https://rarediseases.info.nih.gov/diseases/3314/index
- Catalog of Genes and Diseases from OMIM. GORDON HOLMES SYNDROME; GDHS. Retrieved December 13, 2024 from https://omim.org/entry/212840
- U.S. National Library of Medicine. Gordon Holmes syndrome. Retrieved December 13, 2024 from https://medlineplus.gov/genetics/condition/gordon-holmes-syndrome/
- MalaCards. Gordon Holmes Syndrome (GDHS). Retrieved December 13, 2024 from https://www.malacards.org/card/gordon_holmes_syndrome
- National Institute of Health. Rare case of Gordon Holmes syndrome. Retrieved December 13, 2024 from https://pmc.ncbi.nlm.nih.gov/articles/PMC6040515/
- Orphanet. Cerebellar ataxia-hypogonadism syndrome. Retrieved December 13, 2024 from https://www.orpha.net/en/disease/detail/1173





