Loạn dưỡng cơ liên-quan-LAMA2 (LAMA2-related muscular dystrophy) là bệnh di truyền khiến cơ người bệnh yếu và teo đi, đặc biệt cơ xương—cơ đảm nhiệm chức năng vận động của cơ thể. Bệnh biểu hiện nhiều mức độ khác nhau giữa từng trường hợp, từ dạng nặng khởi phát sớm đến dạng nhẹ khởi phát muộn.
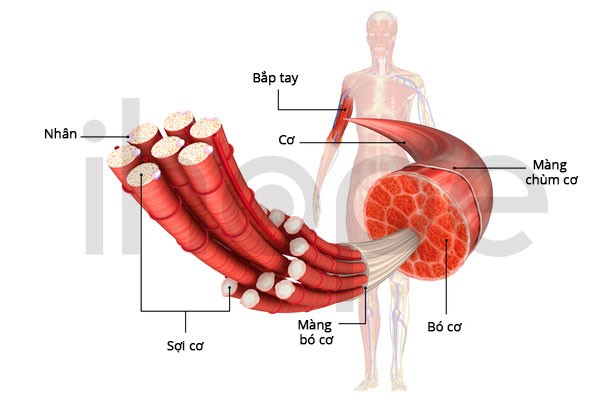
Nguồn: U.S. National Library of Medicine.
Biểu hiện lâm sàng
Dạng khởi phát sớm
Loạn dưỡng cơ liên-quan-LAMA2 dạng khởi phát sớm thuộc nhóm bệnh loạn dưỡng cơ bẩm sinh (loạn dưỡng cơ bẩm sinh loại 1A). Trẻ thường khởi phát bệnh ngay từ khi mới sinh hoặc trong vài tháng đầu đời.
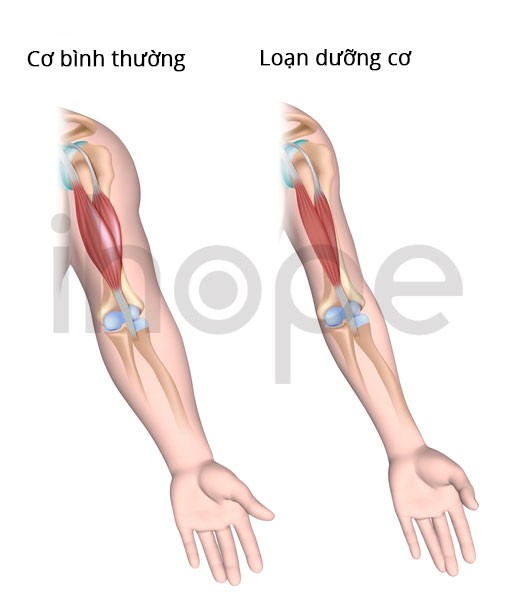
Nguồn: U.S. National Library of Medicine.
Trẻ xuất hiện các dấu hiệu và triệu chứng như:
- Yếu cơ
- Biến dạng khớp
- Trương lực cơ kém
- Cử động tự phát hạn chế
Ngoài ra, cơ mặt và cổ họng suy yếu khiến trẻ ăn uống khó khăn, dẫn đến tình trạng tăng cân chậm và phát triển kém. Ngoài ra, bệnh có thể tác động đến cơ ngực và làm cho trẻ suy hô hấp. Trẻ gặp những vấn đề liên quan đến hô hấp và có tiếng khóc yếu. Do đó, trẻ có nguy cơ nhiễm trùng phổi cao và gặp nguy hiểm đến tính mạng.
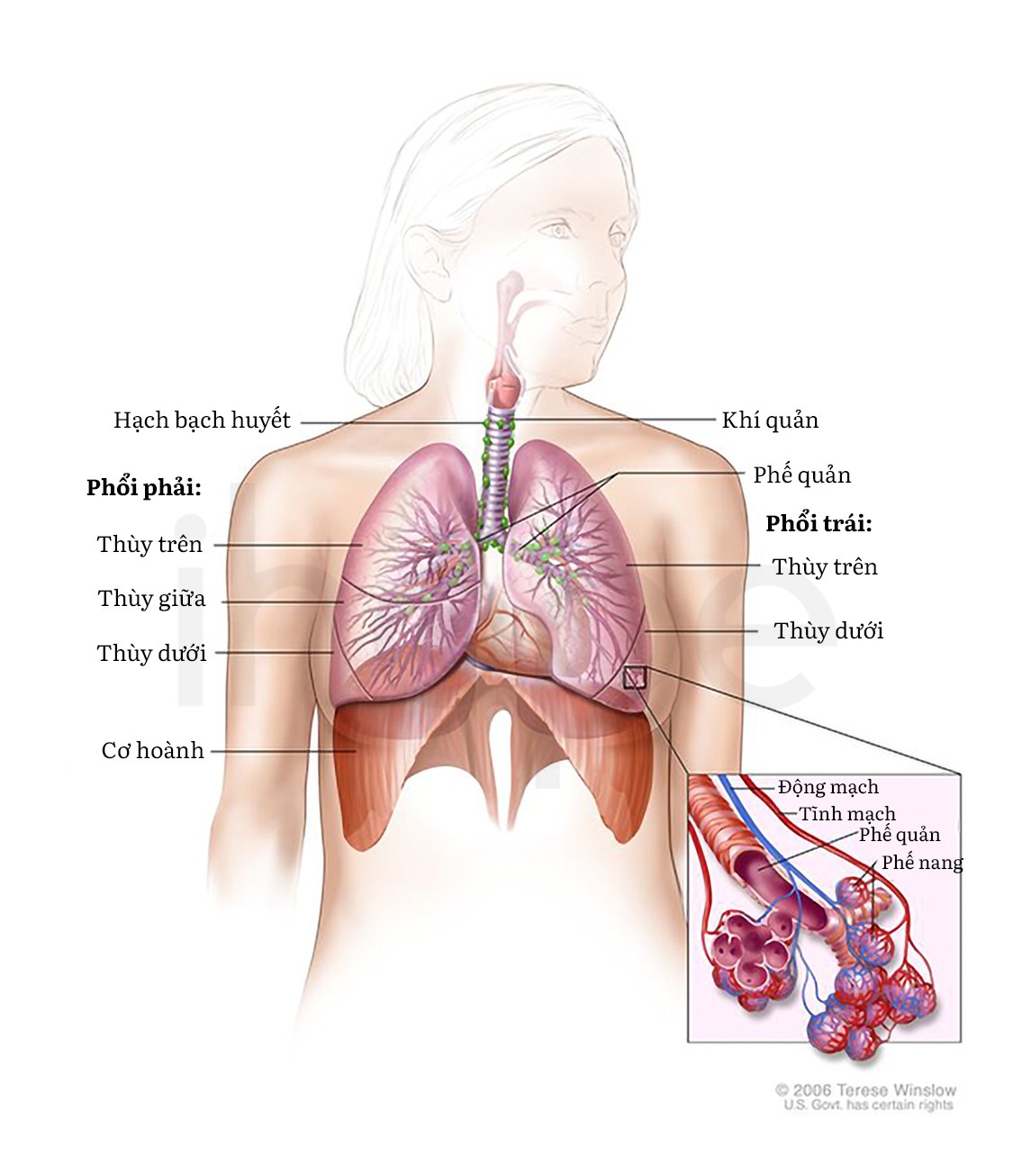
Nguồn: © 2006 Terese Winslow LLC for the National Cancer Institute
Khi trẻ trưởng thành, bệnh tiến triển thành những biểu hiện như:
- Giao tiếp khó khăn
- Vẹo cột sống
- Uỡn cột sống
- Không có khả năng đi lại và vận động
Bệnh ảnh hưởng đến cơ mặt và làm cho lưỡi to , nên trẻ khó nói chuyện. Một phần ba trường hợp loạn dưỡng cơ liên-quan-LAMA2 dạng khởi phát sớm được phát hiện mắc bệnh động kinh. Trường hợp biến chứng tại tim ít phổ biến hơn.
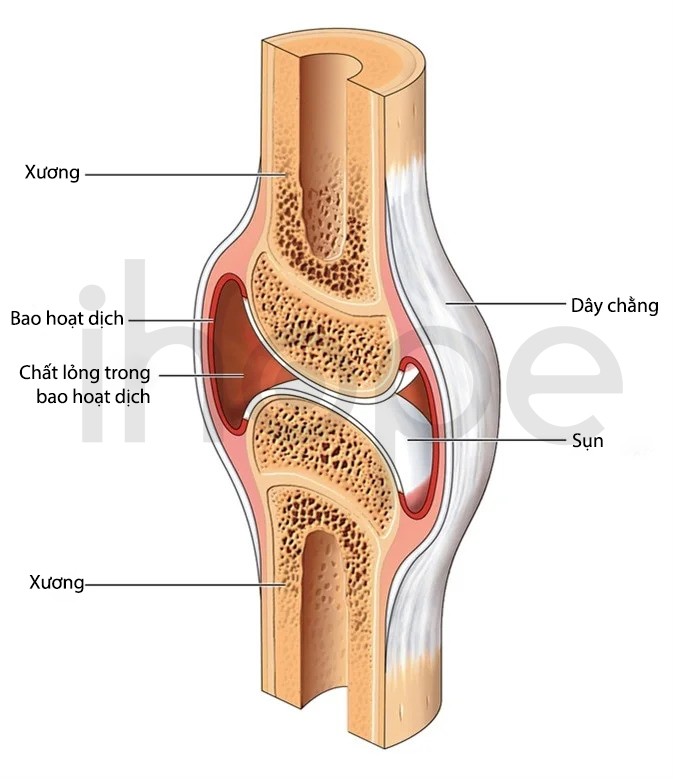
Ảnh: Mặt cắt ngang qua một khớp hoạt dịch điển hình
Nguồn: Blamb/ Shutterstock
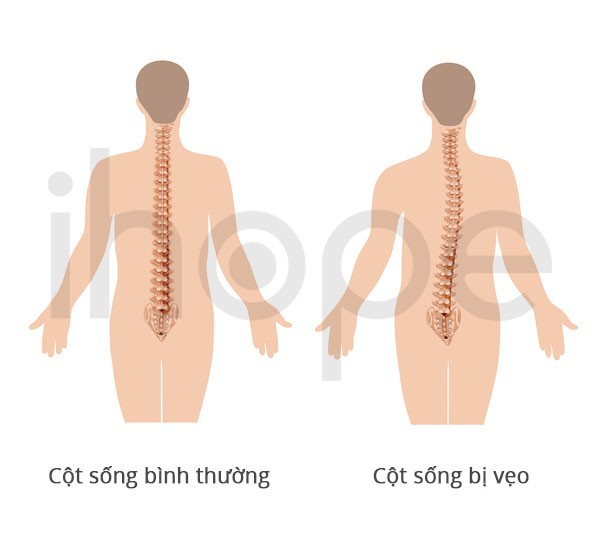
Ảnh: Cột sống bình thường và cột sống bị vẹo
Nguồn: U.S. National Library of Medicine
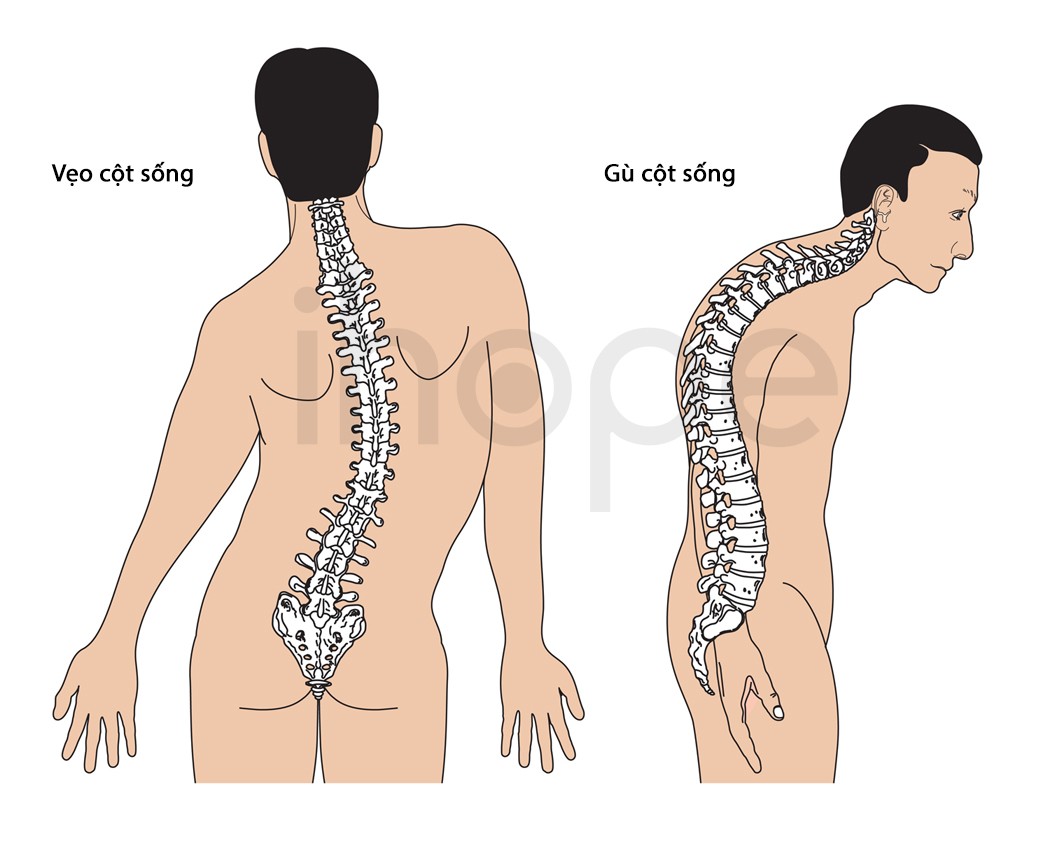
Ảnh: Độ cong bất thường của cột sống
Nguồn: Blamb/Shutterstock.com
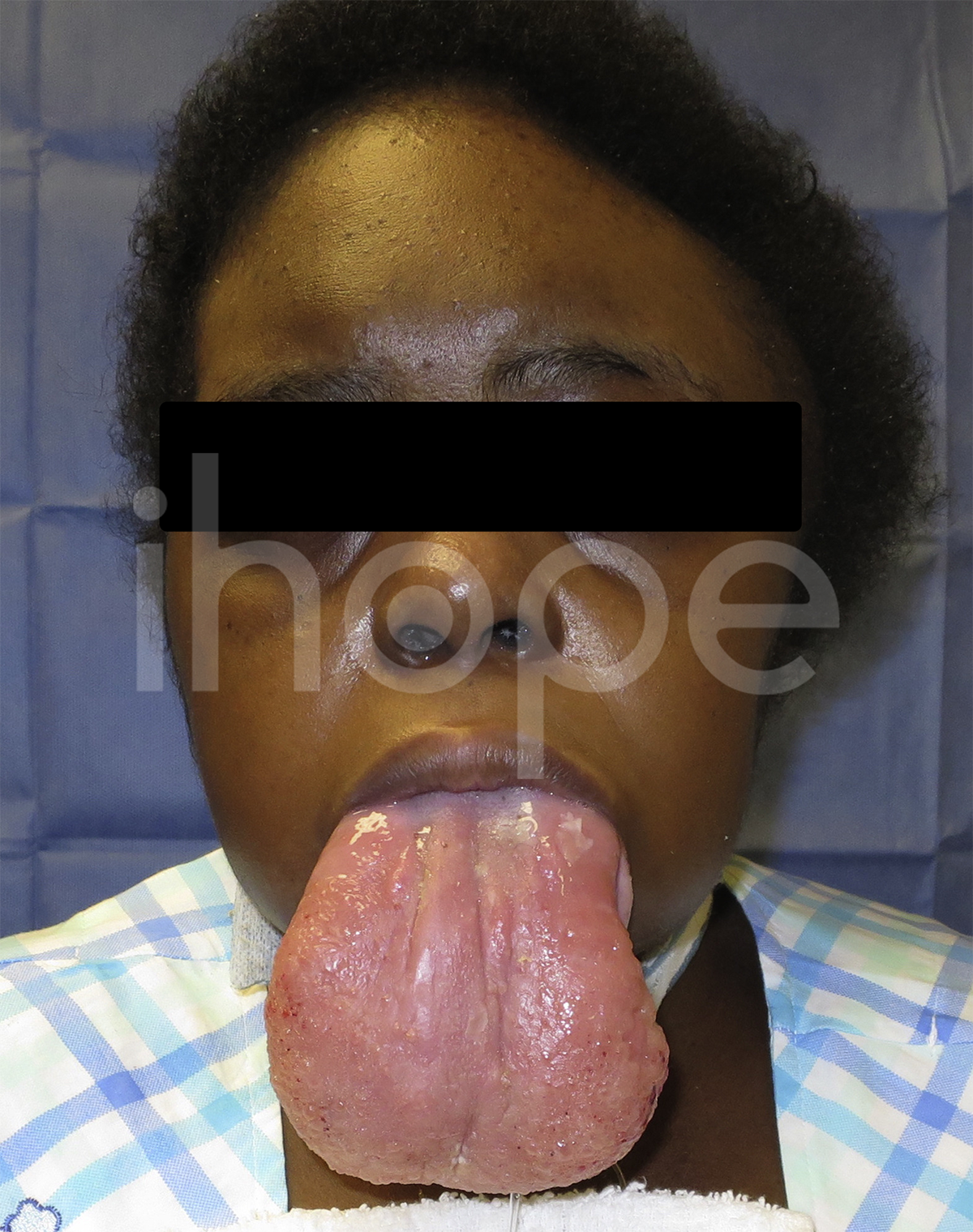
Ảnh: Lưỡi lớn
Nguồn: Elements of Morphology, National Human Genome Research Institute
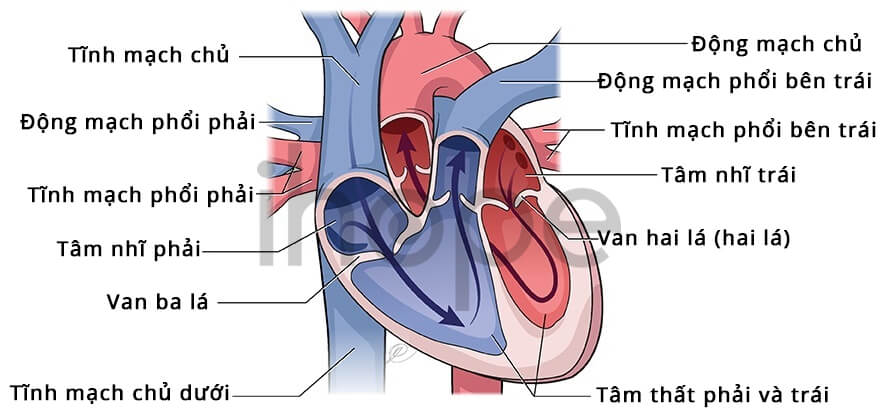
Ảnh: Giải phẫu tim bình thường
Nguồn: U.S National Library of Medicine.
Dạng khởi phát muộn
Loạn dưỡng cơ liên-quan-LAMA2 dạng khởi phát muộn thường bắt đầu tại giai đoạn trẻ nhỏ hoặc khi trưởng thành. Người bệnh có triệu chứng tương tự như nhóm loạn dưỡng cơ limb-girdle. Dạng bệnh này tác động nhiều nhất đến cơ tại các chi gần trung tâm cơ thể như vai, bắp tay, vùng xương chậu và đùi.
Theo thời gian, người bệnh xuất hiện các biểu hiện bao gồm:
- Co rút khớp
- Cứng cột sống
- Vẹo cột sống
- Suy hô hấp
- Chậm phát triển khả năng vận động
Tuy nhiên, người bệnh chỉ giảm khả năng vận động chứ không mất hoàn toàn. Phần lớn bệnh nhân vẫn có khả năng đi lại và leo cầu thang.
Độ phổ biến
Theo thống kê trên toàn thế giới, tỉ lệ mắc loạn dưỡng cơ liên-quan-LAMA2 khoảng 1/50.000–400.000 người. Đây là bệnh phổ biến nhất trong nhóm loạn dưỡng cơ bẩm sinh, nó chiếm từ 30% đến 40% tổng số trường hợp mắc bệnh.
Nguyên nhân
Đột biến gen LAMA2 gây ra bệnh loạn dưỡng cơ liên quan LAMA2. Gen LAMA2 cung cấp hướng dẫn tạo ra tiểu đơn vị alpha-2 trong protein laminin 2 (merosin) và protein laminin 4. Tiểu đơn vị là thành phần cấu tạo nên protein. Họ protein laminin được tạo thành từ ba tiểu đơn vị gồm alpha, beta và gamma. Mỗi tiểu đơn vị có nhiều dạng cấu trúc khác nhau, thông tin của mỗi dạng này được các gen tương ứng quy định tạo ra.
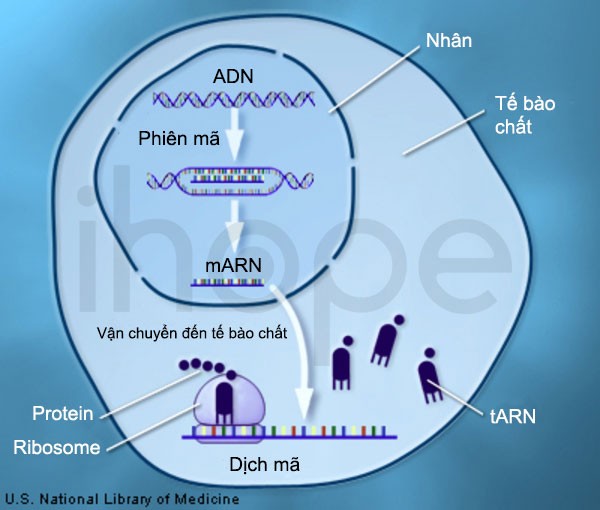
Nguồn: U.S. National Library of Medicine
Laminin tồn tại trong chất nền ngoại bào—mạng lưới phức tạp gồm protein và nhiều phân tử khác được hình thành tại khoảng trống giữa những tế bào. Protein laminin 2 và protein laminin 4 đặc biệt quan trọng đối với cơ dùng để vận động như cơ xương. Laminin liên kết với các protein khác trong chất nền ngoại bào và trong màng tế bào cơ, từ đó tính ổn định của sợi cơ được duy trì.
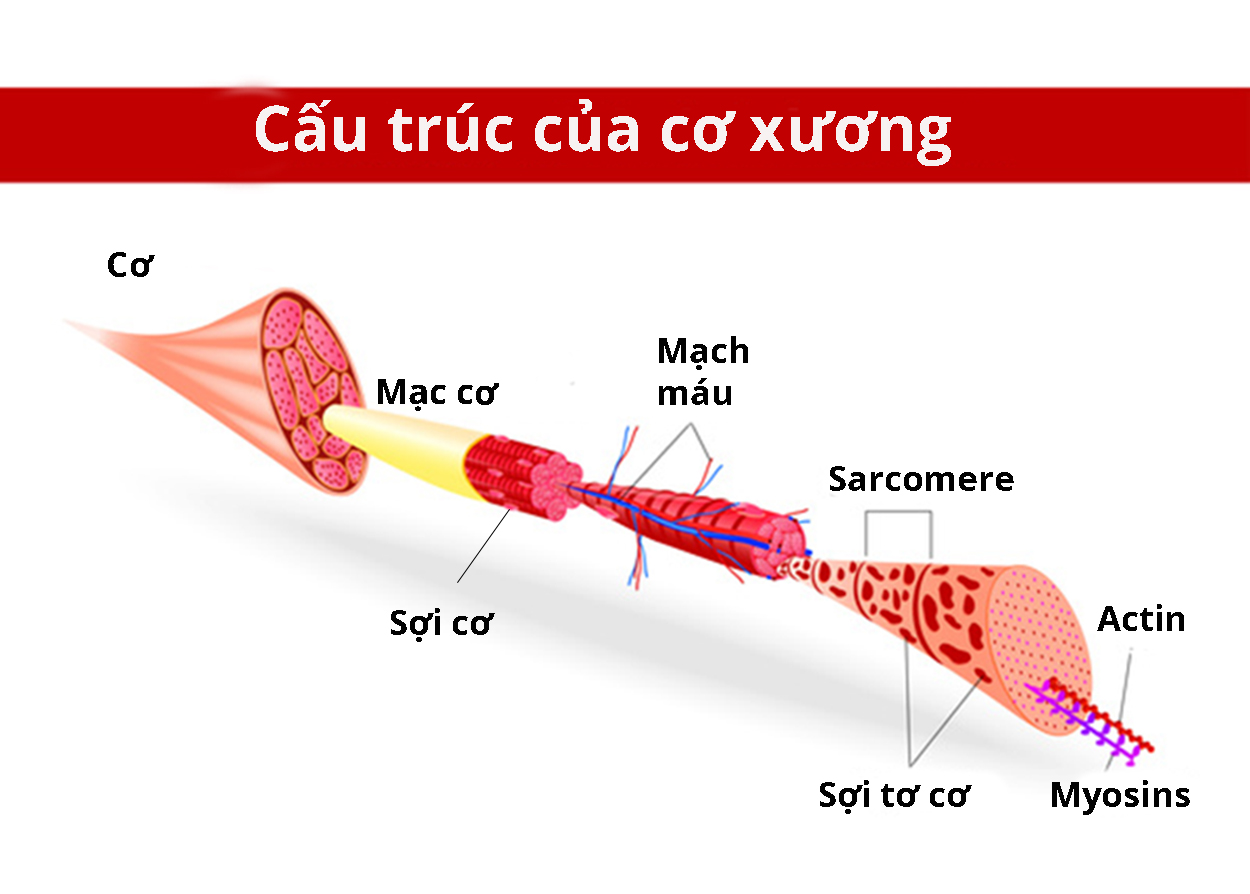
Nguồn: Designua/Shutterstock.com
Gen LAMA2 đột biến làm giảm số lượng tiểu đơn vị laminin alpha-2 được tạo ra, từ đó người bệnh biểu hiện dạng nhẹ. Đối với dạng nặng, người bệnh mất hoàn toàn tiểu đơn vị laminin alpha-2. Thiếu hoặc không có tiểu đơn vị laminin alpha-2 dẫn đến thiếu laminin 2 và laminin 4 tương ứng. Do đó, mô cơ giảm sức mạnh, mất ổn định rồi gây ra triệu chứng bệnh.
Chẩn đoán
Loạn dưỡng cơ liên-quan-LAMA2 có thể được chẩn đoán dựa trên biểu hiện lâm sàng của người bệnh kết hợp nhiều xét nghiệm chuyên biệt để xác nhận kết quả chẩn đoán. Bệnh nhân nên thực hiện chẩn đoán phân biệt để tránh nhầm lẫn với những bệnh lý khác có triệu chứng tương tự như hội chứng nhược cơ bẩm sinh hay teo cơ cột sống. Ngoài ra, bác sĩ có thể chỉ định người bệnh làm xét nghiệm di truyền để phát hiện gen LAMA2 đột biến.
Những phương pháp dùng để chẩn đoán loạn dưỡng cơ liên quan LAMA2 bao gồm:
- Chụp MRI não
- Xét nghiệm máu kiểm tra nồng độ creatine kinase trong huyết thanh
- Nhuộm hóa mô miễn dịch từ mẫu cơ hoặc da sinh thiết
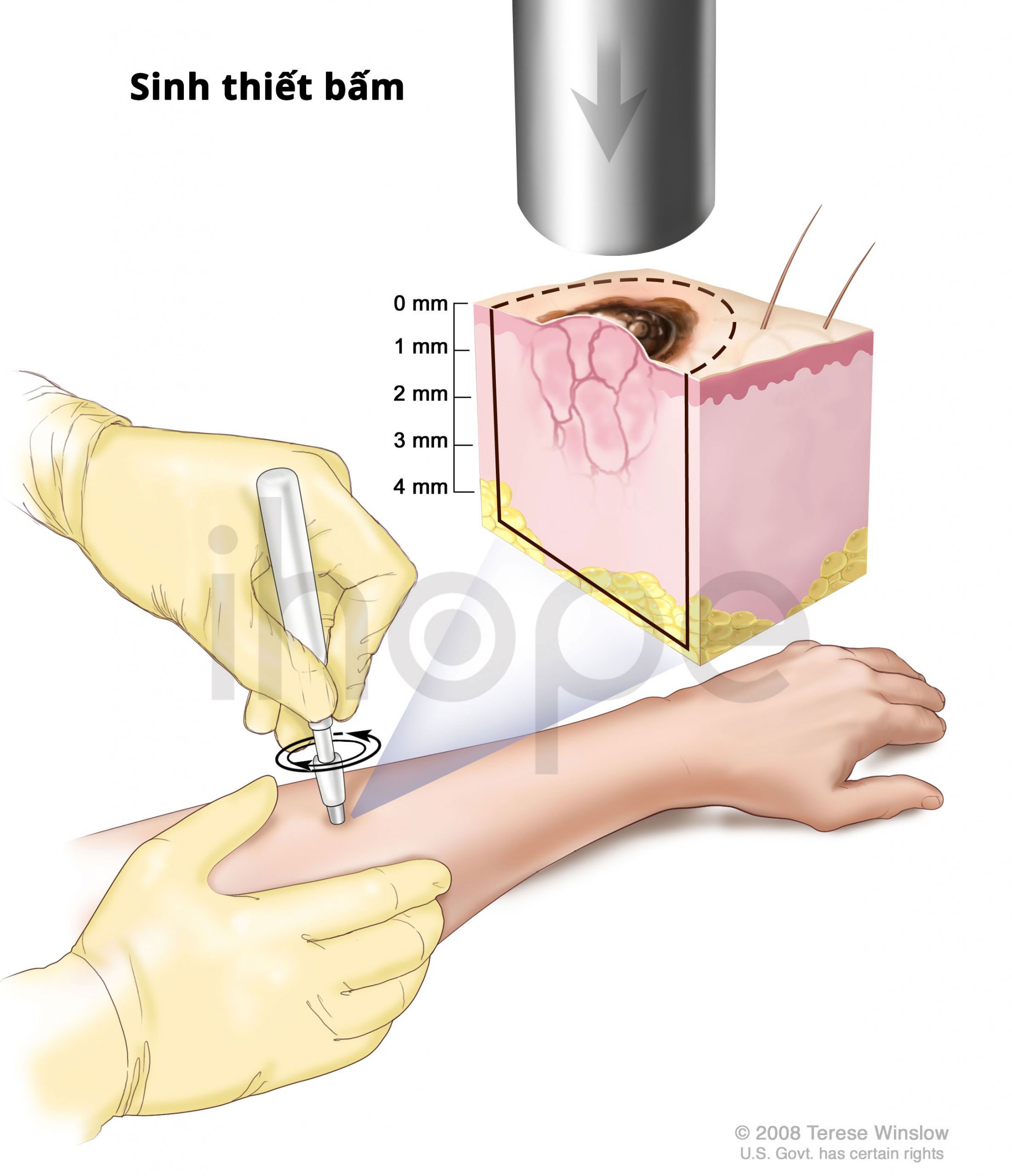
Nguồn: Terese Winslow
Điều trị
Hiện nay, chưa có phương pháp điều trị hoàn toàn bệnh loạn dưỡng cơ liên-quan-LAMA2. Các liệu pháp được cá nhân hóa dựa trên biểu hiện, triệu chứng và mức độ nghiêm trọng nhằm đảm bảo liệu pháp điều trị toàn diện và phù hợp với từng bệnh nhân. Điều trị chủ yếu nhằm giảm nhẹ triệu chứng và cải thiện chất lượng đời sống cho người bệnh.
Bệnh loạn dưỡng cơ liên-quan-LAMA2 nên thực hiện điều trị phối hợp nhiều lĩnh vực như:
- Phổi
- Tim mạch
- Thần kinh
- Chăm sóc tiêu hóa và dinh dưỡng
- Chỉnh hình và phục hồi chức năng
Dạng di truyền
Loạn dưỡng cơ liên-quan-LAMA2 di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh lặn trên nhiễm sắc thể thường sẽ có bố và mẹ mang một bản sao của gen đột biến, nhưng họ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Loạn dưỡng cơ liên-quan-LAMA2 di truyền lặn do đột biến gen LAMA2, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kỳ.
Các tên gọi khác
- MDC1A
- LAMA2 MD
- Laminin alpha 2 deficiency
- Merosin-deficient muscular dystrophy
- Muscular dystrophy due to LAMA2 deficiency
- Laminin alpha-2 deficient muscular dystrophy
References
- Genetic Testing Information. Congenital muscular dystrophy due to partial LAMA2 deficiency. Retrieved November 28, 2023 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C1842898/
- Genetic and Rare Diseases Information Center. Laminin subunit alpha 2-related congenital muscular dystrophy. Retrieved November 28, 2023 from https://rarediseases.info.nih.gov/diseases/3843/congenital-muscular-dystrophy-type-1a
- Catalog of Genes and Diseases from OMIM. MUSCULAR DYSTROPHY, CONGENITAL MEROSIN-DEFICIENT, 1A; MDC1A. Retrieved November 28, 2023 from https://omim.org/entry/607855
- U.S National Library of Medicine. LAMA2-related muscular dystrophy. Retrieved November 28, 2023 from https://medlineplus.gov/genetics/condition/lama2-related-muscular-dystrophy/
- Frontiers. LAMA2-Related Dystrophies: Clinical Phenotypes, Disease Biomarkers, and Clinical Trial Readiness. Retrieved November 28, 2023 from https://www.frontiersin.org/articles/10.3389/fnmol.2020.00123/full
- MalaCards. Lama2 Muscular Dystrophy. Retrieved November 28, 2023 from https://www.malacards.org/card/lama2_muscular_dystrophy
- National Cancer Institute. LAMA2-related muscular dystrophy. Retrieved November 28, 2023 from https://moleculartargets.ccdi.cancer.gov/disease/MONDO_0100228/associations
- National Institute of Health. LAMA2-Related Muscular Dystrophy Across the Life Span. Retrieved November 28, 2023 from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10356133/
- Orphanet. Laminin subunit alpha 2-related muscular dystrophy. Retrieved November 28, 2023 from https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=207094





