
Loạn dưỡng tế bào hình nón và hình que (cone-rod dystrophy) là nhóm các rối loạn ảnh hưởng đến võng mạc, bao gồm tế bào hình nón và tế bào hình que giữ chức năng nhạy cảm với ánh sáng phía sau mắt. Triệu chứng của bệnh tiến triển nặng hơn theo thời gian, gây nhiều khó khăn trong đời sống của người bệnh.
Nguồn: U.S. National Library of Medicine
Biểu hiện lâm sàng
Người bệnh mất thị lực khi các tế bào cảm nhận ánh sáng của võng mạc bị thoái hóa. Đầu tiên, mắt của họ giảm độ sắc nét và tăng nhạy cảm với ánh sáng, sau đó khả năng nhận biết màu suy giảm, điểm mù xuất hiện tại trung tâm của trường thị giác và thị lực ngoại vi mất đi. Theo thời gian, người bệnh phát triển chứng quáng gà, nên họ gặp khó khăn khi di chuyển.
Giảm thị lực khiến đọc chữ trở nên khó khăn, đa số người bệnh bị mù về mặt pháp lý tại tuổi trung niên. Khi bệnh tiến triển, cá nhân có thể phát triển các chuyển động mắt không tự nguyện (rung giật nhãn cầu).
Bệnh được chia thành khoảng 30 loại dựa trên nguyên nhân và kiểu di truyền. Ngoài ra, bệnh có thể xảy ra đơn lẻ hoặc thuộc hội chứng khác.
Độ phổ biến
Tỷ lệ mắc bệnh ước tính khoảng 1/30.000 - 1/40.000 người.
Nguyên nhân
Đột biến hơn 30 gen gây loạn dưỡng tế bào hình nón và hình que, trong đó khoảng 20 gen di truyền kiểu lặn trên nhiễm sắc thể thường.
Đột biến một số gen phổ biến đã được xác định, bao gồm:
- 30-60% các trường hợp do đột biến gen ABCA4 và di truyền lặn trên nhiễm sắc thể thường.
- Khoảng 10 đột biến gen gây bệnh di truyền trội trên nhiễm sắc thể thường, trong đó đột biến gen GUCY2D và CRX chiếm 50% trường hợp.
- Đột biến gen RPGR có kiểu di truyền liên kết X.
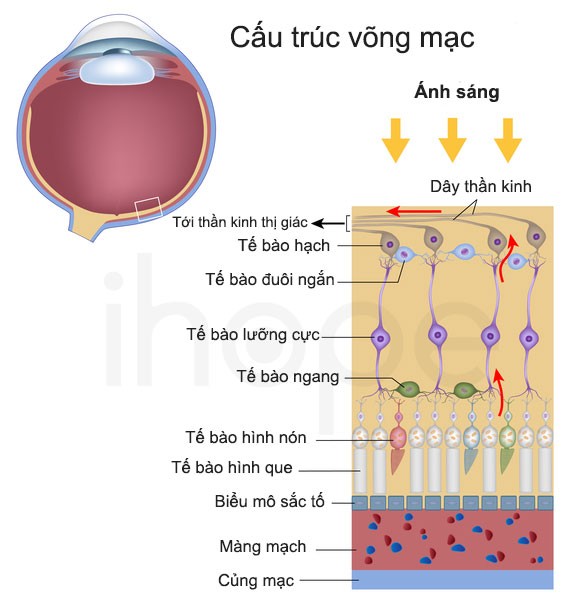
Võng mạc là lớp mô nằm sau mắt, nó chứa các tế bào cảm quang gồm tế bào hình nón và tế bào hình que. Các tế bào này phát hiện và phản ứng với kích thích từ ánh sáng. Tế bào hình que có khả năng cảm nhận ánh sáng trong điều kiện ánh sáng yếu (ban đêm). Trong khi đó, tế bào hình nón cảm nhận ánh sáng mạnh giúp mắt có thể nhìn thấy và phân biệt màu sắc. Những gen trên quy định cấu trúc, chức năng của tế bào nón và hình que.
Đột biến trong các gen liên quan làm thoái hóa tế bào cảm quang của võng mạc. Đầu tiên, tế bào hình nón bị phá vỡ khiến người bệnh suy giảm thị lực và mất khả năng phân biệt màu sắc. Sau đó, tế bào hình que bị phân hủy, người bệnh bị giảm khả năng nhìn ban đêm.
Chẩn đoán
Chẩn đoán bệnh gồm nhiều bước khác nhau nhằm xác định các triệu chứng của bệnh và đánh giá tình trạng mắt.
Bác sĩ sẽ tiến hành một số xét nghiệm bao gồm:
- Kiểm tra triệu chứng như khả năng nhĩn rõ, nhận diện màu sắc, nhạy cảm với ánh sáng hoặc các triệu chứng liên quan khác
- Kiểm tra mắt bằng các thiết bị như đèn kính, mát quét võng mạc, máy siêu âm mắt hoặc thử nghiệm trường thị lực
- Kiểm tra chức năng thị lực bằng thử nghiệm nhìn chữ
- Chụp ảnh võng mạc xác định tổn thương của võng mạc
Kết quả xét nghiệm có thể không thấy bất thường trong giai đoạn đầu. Tuy nhiên, bệnh chuyển biến nặng theo thời gian, do đó cần lặp lại chẩn đoán sau lần đầu từ 1-2 năm. Kết quả xét nghiệm tại giai đoạn muộn có thể bao gồm thoái hóa lớp tế bào biểu mô sắc tố, suy giảm tiểu động mạch. Ngoài ra, xét nghiệm di truyền giúp phát hiện các đột biến gen liên quan để chẩn đoán.
Điều trị
Hiện nay chưa có phương pháp điều trị hoàn toàn loạn dưỡng tế bào hình nón và hình que. Mục tiêu điều trị chủ yếu kiểm soát triệu chứng và ngăn ngừa bệnh tiến triển.
Các phương pháp điều trị bao gồm:
- Bổ sung vitamin A và sử dụng các thuốc chống oxy hóa
- Dùng kính áp tròng hỗ trợ thị lực
- Phẫu thuật cấy ghép giác mạc
Dạng di truyền
Loạn dưỡng tế bào hình nón và hình que có cơ chế di truyền phức tạp.
Trường hợp bệnh di truyền kiểu lặn nhiễm sắc thể thường. Bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh lặn trên nhiễm sắc thể thường sẽ có bố và mẹ mang một bản sao của gen đột biến, nhưng bố mẹ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Bệnh di truyền trội trên nhiễm sắc thể ít gặp. Do đó, một bản sao của gen trong mỗi tế bào đủ để gây ra bệnh.

Nguồn: U.S. National Library of Medicine
Ngoài ra, bệnh có thể di truyền lặn liên kết nhiễm sắc thể X nhưng thường hiếm gặp. Nam giới chỉ có một nhiễm sắc thể X nên một bản sao gen đột biến trong mỗi tế bào đủ gây ra bệnh. Nữ giới mang đột biến trong một bản sao của gen đột biến thường không bị ảnh hưởng. Một số người bị đột biến cả hai bản sao của gen có các triệu chứng nhẹ như chuột rút, run, điều này có thể do nồng độ androgen thấp hơn. Một đặc điểm của bệnh di truyền liên kết X là người cha không thể truyền các đặc điểm này cho con trai của họ.
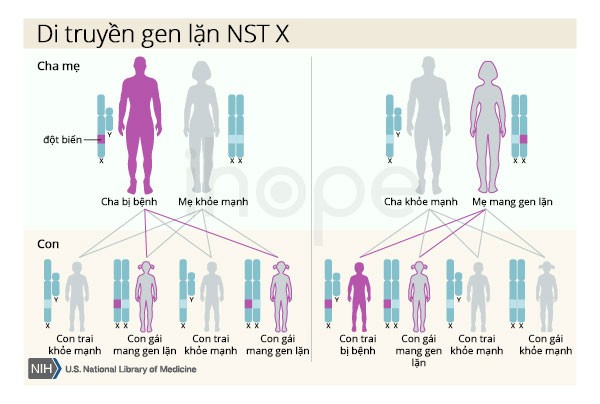
Nguồn: U.S. National Library of Medicine
Phòng ngừa
Bệnh có cơ chế di truyền phức tạp. Người mắc bệnh nên được khám sức khỏe kỹ nhằm tìm ra dạng di truyền của bệnh, từ đó có phương pháp phòng ngừa phù hợp.
Trường hợp bệnh di truyền lặn trên nhiễm sắc thể thường, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh. Ngoài ra, bệnh di truyền liên kết X phức tạp nên khó phát hiện ở những người phụ nữ mang gen bệnh cho đến khi sinh con. Để chủ động phòng ngừa, người mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con.
Bệnh có thể di truyền trội nhiễm sắc thể thường. Nếu cha hoặc mẹ mắc bệnh, con sinh ra sẽ có 50% khả năng di truyền bệnh. Do đó, để bảo đảm 100% khả năng con không bị bệnh, cha mẹ có thể chọn phương pháp thụ tinh nhân tạo IVF và sàng lọc phôi PGS/PGD.
Các thành viên trong gia đình nên khám sức khỏe và tầm soát bệnh định kỳ nếu có thành viên mắc bệnh. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh.
Các tên gọi khác
- Cone-rod degeneration
- Cone-rod retinal dystrophy
- CORD
- CRD
- Retinal cone-rod dystrophy
- Tapetoretinal degeneration
References
- Genetic Testing Information. Cone-rod dystrophy. Retrieved March 21, 2023 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C4085590/
- Genetic and Rare Diseases Information Center. Cone-rod dystrophy. Retrieved March 21, 2023 from https://rarediseases.info.nih.gov/diseases/10790/cone-rod-dystrophy/
- Catalog of Genes and Diseases from OMIM. CONE-ROD DYSTROPHY 1. Retrieved March 21, 2023 from https://omim.org/entry/600624
- U.S National Library of Medicine. Cone-rod dystrophy. Retrieved March 21, 2023 from https://medlineplus.gov/genetics/condition/cone-rod-dystrophy/
- Frontiers. Autosomal Recessive Rod-Cone Dystrophy Associated With Compound Heterozygous Variants in ARL3 Gene. Retrieved March 21, 2023 from https://www.frontiersin.org/articles/10.3389/fcell.2021.635424/full
- MalaCards. Cone-Rod Dystrophy 5 (CORD5). Retrieved March 21, 2023 from https://www.malacards.org/card/cone_rod_dystrophy_5
- National Organization for Rare Disorders. Cone Dystrophy. Retrieved March 21, 2023 from https://rarediseases.org/rare-diseases/cone-dystrophy/
- Orphanet. Cone rod dystrophy. Retrieved March 21, 2023 from https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=GB&Expert=1872
- Ophthalmology Retina. The Natural History of Leber Congenital Amaurosis and Cone–Rod Dystrophy Associated with Variants in the GUCY2D Gene. Retrieved March 21, 2023 from https://www.ophthalmologyretina.org/article/S2468-6530(22)00107-5/fulltext





