Tăng tyrosine máu loại I (tyrosinemia type 1) thuộc nhóm bệnh rối loạn chuyển hóa axit amin, người bệnh không thể phân hủy tyrosine—một loại axit amin được hấp thụ từ thực phẩm. Quá trình phân hủy tyrosine diễn ra qua nhiều bước, trong đó mỗi bước cần một enzyme chuyên biệt. Dựa vào loại enzyme bị thiếu, người ta chia bệnh thành ba dạng bao gồm tăng tyrosine máu loại I, loại II và loại III.
Nguồn: Darryl Leja, NHGRI
Đối với người bệnh thuộc dạng tăng tyrosine máu loại I, enzyme fumarylacetoacetate hydrolase (FAH) không hoạt động nên lượng tyrosine cao bất thường trong gan, thận và hệ thần kinh trung ương. Do đó, người bệnh bị ra suy gan nghiêm trọng cũng như gặp các vấn đề sức khỏe khác.
Biểu hiện lâm sàng
Đối với từng cá thể, các triệu chứng liên quan đến tăng tyrosine máu loại I thường rất khác nhau. Bệnh có hai dạng bao gồm cấp tính và mãn tính.
Dạng cấp tính
Dạng cấp tính có mức độ nặng hơn và khởi phát sớm ngay từ khi trẻ mới sinh. Dạng này cũng xảy ra phổ biến hơn so với dạng mãn tính.
Một số triệu chứng đầu tiên của trẻ bệnh dạng cấp tính gồm:
- Nôn mửa
- Dễ cáu gắt
- Tăng cân kém
- Ngủ nhiều bất thường
- Tiêu chảy và phân lẫn máu
- Da hoặc nước tiểu có mùi giống bắp cải
Ngoài ra, tăng tyrosine máu loại I còn ảnh hưởng đến gan , do đó người bệnh bị suy gan với các biểu hiện như:
- Gan to
- Vàng da
- Phù nề bụng và chân
- Dễ chảy máu và bầm tím
Ngoài ra, bệnh cũng tác động đến chức năng thận nên trẻ xuất hiện tình trạng loãng xương , còi xương cũng như chậm biết đi hơn so với trẻ cũng lứa tuổi. Trường hợp gặp vấn đề nghiêm trọng về gan và thận, trẻ sơ sinh có thể tử vong nếu không được điều trị kịp thời.
Nguồn: National Cancer Institue
Dấu hiệu khác của bệnh tyrosine máu loại I bao gồm:
- Co giật
- Hôn mê
- Lách to
- Nhịp tim nhanh
- Chân tay đau, tê yếu
- Rối loạn đông máu
- Bệnh liên quan đến hô hấp
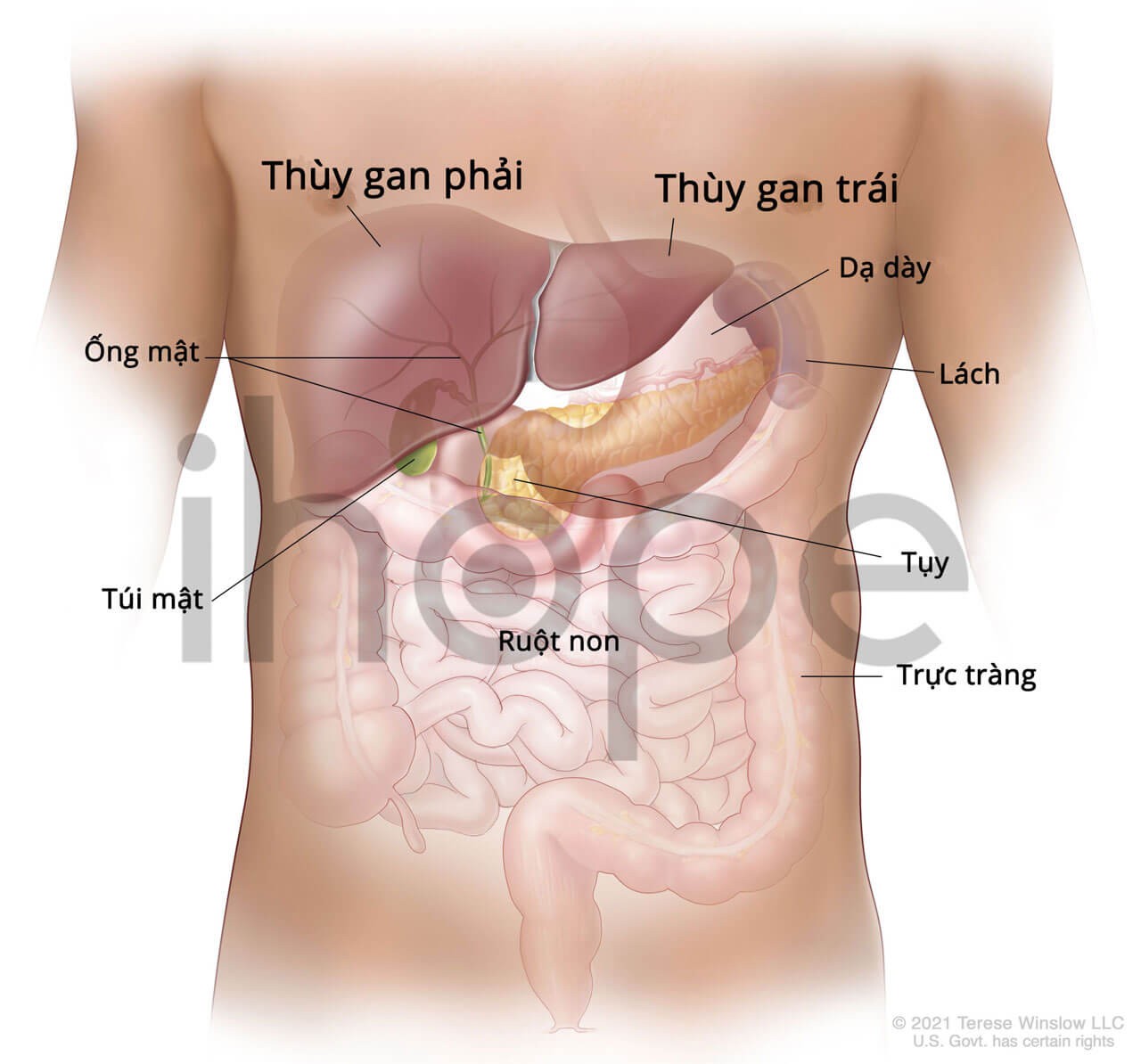
Ảnh: giải phẫu gan người
Nguồn: Terese Winslow LLC[/caption]
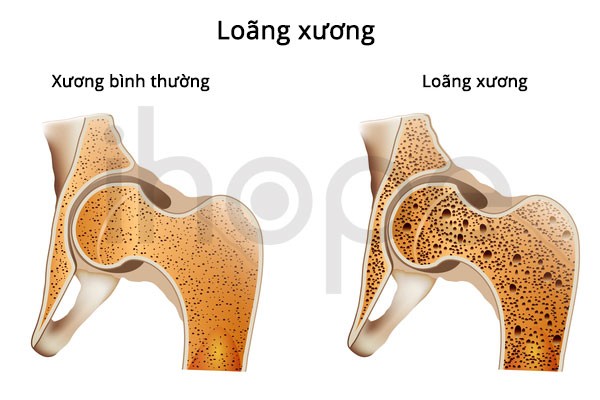
Ảnh: Loãng xương
Nguồn: U.S. National Library of Medicine
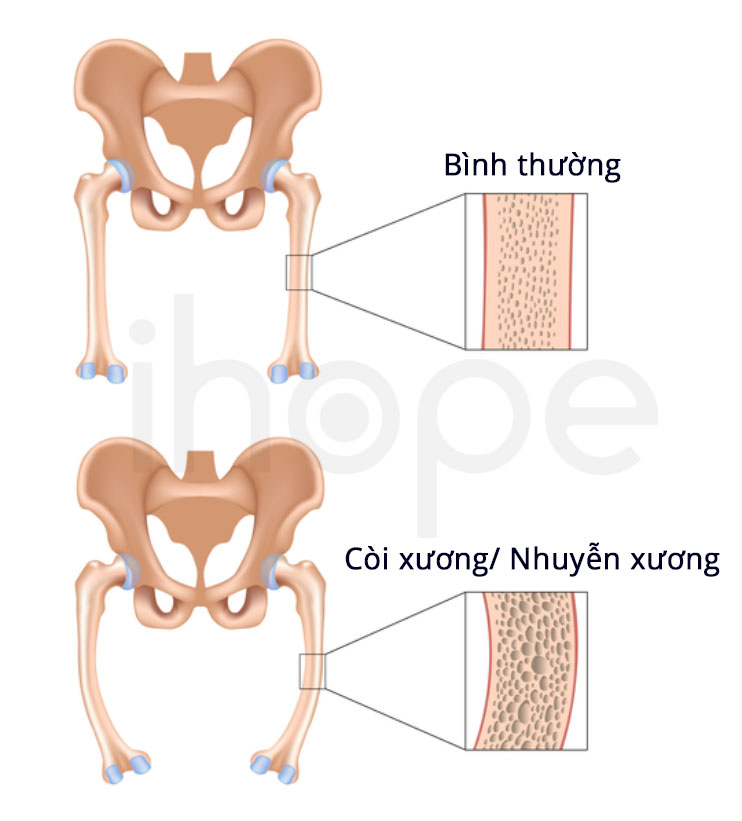
Ảnh: Bệnh còi xương
Nguồn: Alila Medical Media/Shutterstock.com
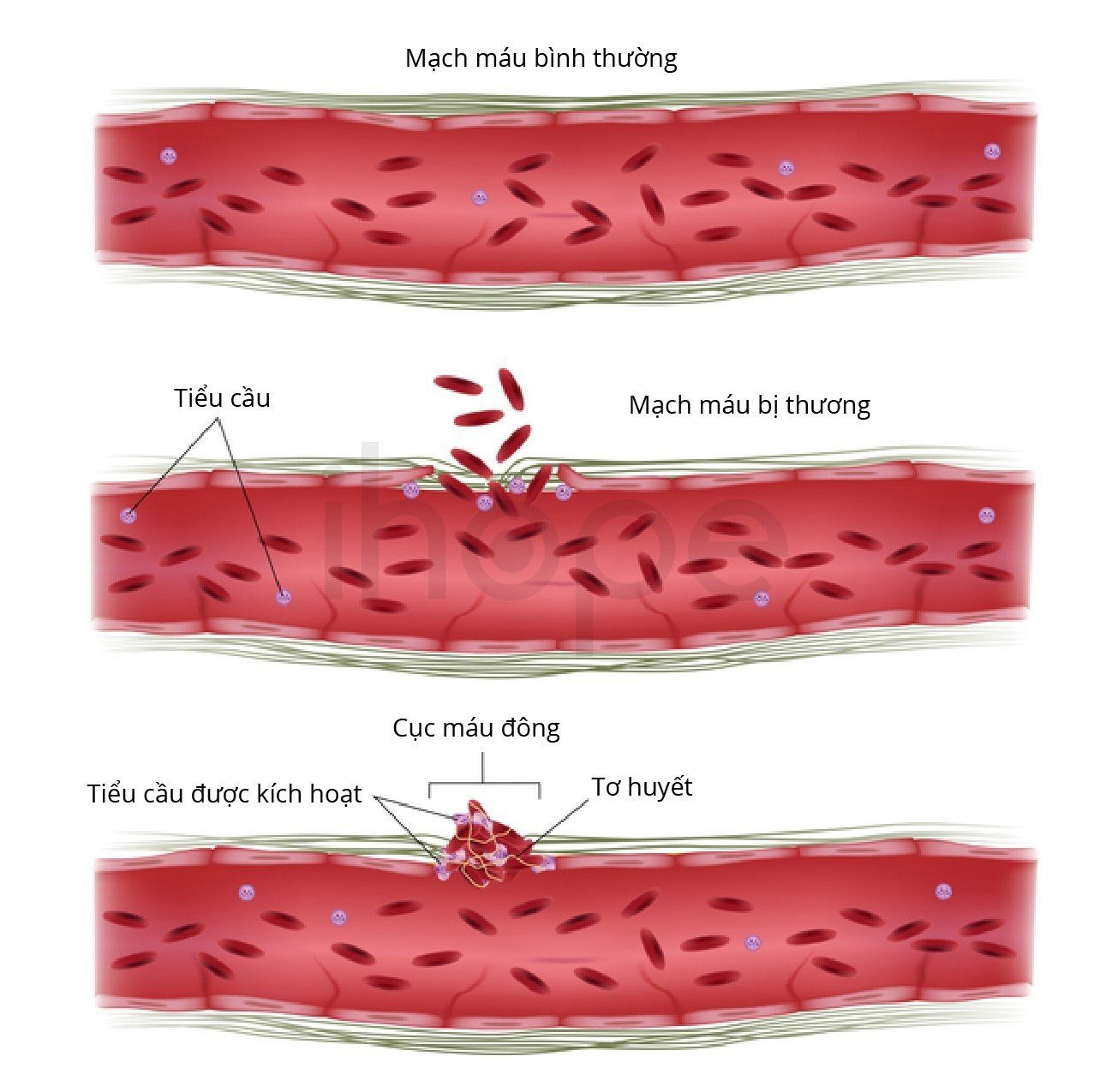
Ảnh: Hình thành cục máu đông
Nguồn: Medlineplus.gov
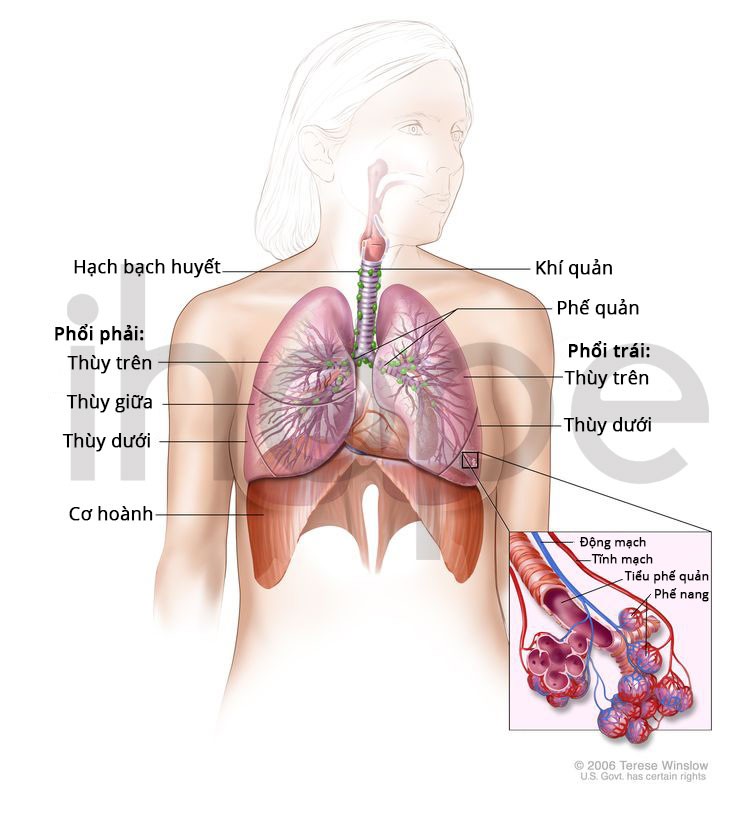
Ảnh: Cấu trúc hệ hô hấp
Nguồn: National Cancer Institute
Dạng mãn tính
Đối với dạng mãn tính, bệnh có mức độ biểu hiện nhẹ và ít phổ biến. Trẻ bệnh dạng này thường khởi phát triệu chứng sau hai tháng tuổi hoặc biểu hiện rõ ràng sau sáu tháng tuổi. Dấu hiệu đầu tiên có thể bao gồm khó tăng cân, nôn mửa và tiêu chảy. Theo thời gian, triệu chứng trở nên nghiêm trọng hơn, từ đó nhiều cơ quan như gan, thận, tim và hệ thần kinh bị ảnh hưởng.
Triệu chứng bao gồm:
- Gan: nhiều nốt xơ gan (nodular cirrhosis) hình thành nên bệnh nhân có thể bị suy gan hoặc ung thư gan trước 10 tuổi nếu không được điều trị. Ngoài ra, người bệnh có nguy cơ cao mắc ung thư biểu mô tế bào gan.
- Thận: hội chứng Fanconi làm rối loạn chức năng thận và ảnh hưởng đến cấu trúc xương . Hội chứng này cũng liên quan đến các biểu hiện như nôn mửa, mất nước, suy nhược cơ thể và sốt.
- Tim: tim chịu tác động dẫn đến bệnh cơ tim phì đại
- Thần kinh: khoảng 40% trẻ sơ sinh mắc bệnh đa dây thần kinh sau khi trải qua một đợt nhiễm trùng nhẹ. Hệ thần kinh bị thương tổn khiến trẻ có những dấu hiệu như tăng trương lực cơ, đau chân, liệt ruột và tăng huyết áp. Một số người bệnh có hành vi tự hại bản thân như cắn lưỡi hoặc nghiến răng.
Nguồn: Cleveland Clinic
Nguồn: U.S. National Library of Medicine
Nguồn: Encyclopædia Britannica, Inc.
Ảnh: Giải phẫu xương người
Nguồn: Terese Winslow LLC
Độ phổ biến
Theo số liệu thống kê, tỉ lệ trẻ mắc bệnh tăng tyrosine máu loại I khoảng 1/100.000–120.000 trường hợp. Tuy nhiên, tỉ lệ này giảm tại nhiều quốc gia thực hiện chương trình sàng lọc sơ sinh bắt buộc, tỉ lệ mắc bệnh chỉ còn 1/100–150 trẻ.
Ngoài ra, bệnh phổ biến hơn đối với người Canada gốc Pháp. Tại Quebec, tỉ lệ mắc bệnh là 1/16.000; tại vùng Saguenay-Lac St. Jean thuộc Quebec, tỉ lệ này là 1/1.864.
Nguyên nhân
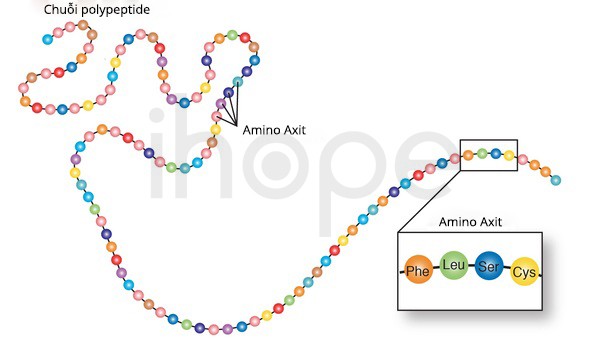
Đột biến gen FAH gây ra bệnh tăng tyrosine máu loại I. Gen này cung cấp hướng dẫn tạp ra enzyme fumarylacetoacetate hydrolase. Enzyme fumarylacetoacetate hydrolase xếp vị trí cuối cùng trong nhóm năm enzyme thực hiện chuỗi phản ứng để phân hủy axit amin tyrosine. Để cơ thể có thể hấp thụ được protein từ thực phẩm, protein được phân giải thành các phần nhỏ hơn là axit amin. Sau đó, enzyme đặc hiệu sẽ chuyển hóa axit amin để cơ thể có thể sử dụng.
Cụ thể hơn, fumarylacetoacetate hydrolase chuyển đổi fumarylacetoacetate—sản phẩm phụ của tyrosine— thành các phân tử nhỏ hơn. Sau đó, cơ thể đào thải những phân tử này qua thận hoặc sử dụng chúng để sản xuất năng lượng hay tái tạo thành các chất khác.
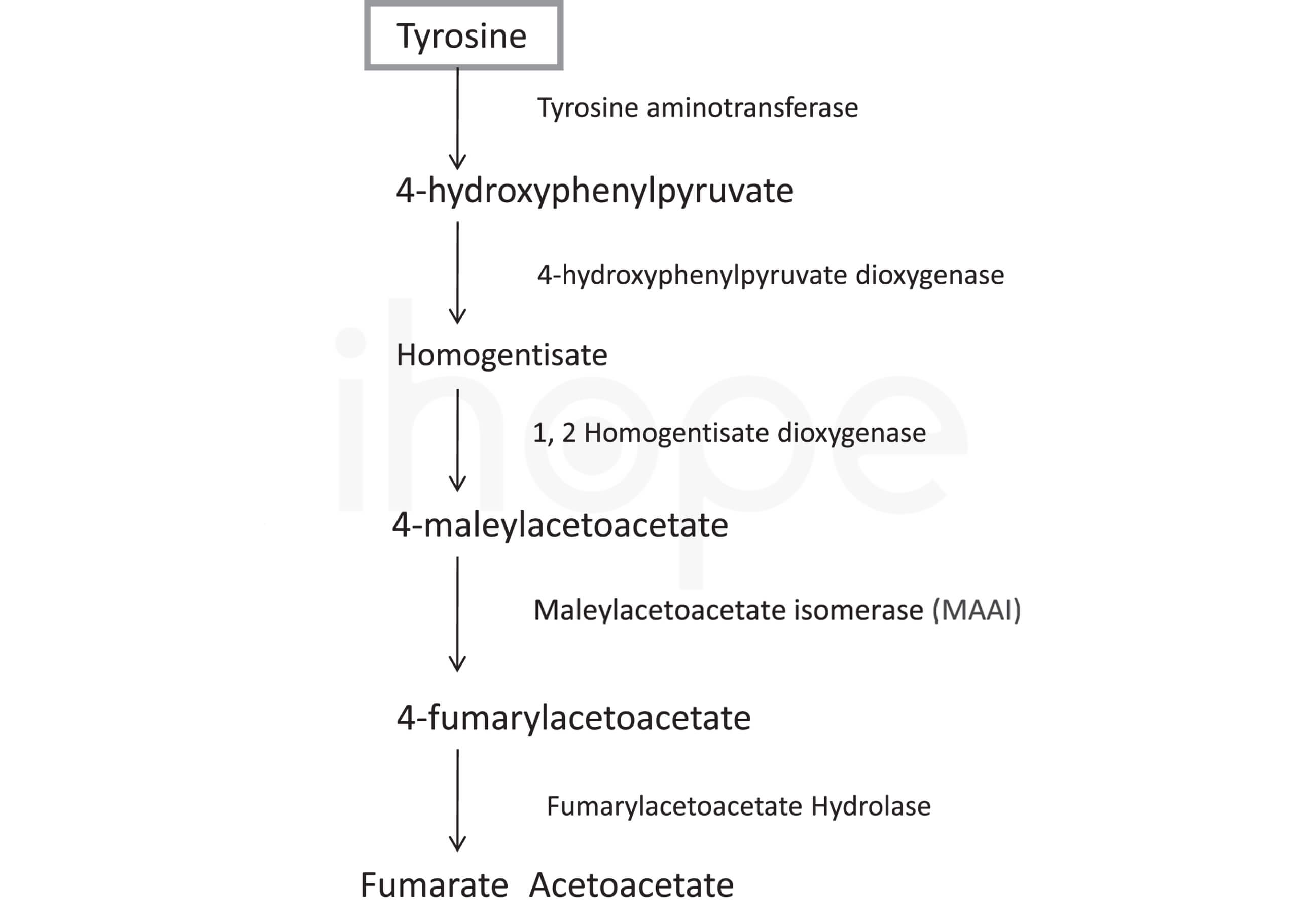
Nguồn: IOS Press
Gen FAH đột biến khiến cơ thể sản xuất ít enzyme hơn bình thường hoặc enzyme tạo ra bị mất chức năng nên chuỗi phản ứng phân giải tyrosine bị gían đoạn. Do đó, tyrosine và các chất chuyển hóa của nó như succanylacetione tích tụ nhiều bất thường trong máu. Nếu nồng đồ những chất này trong máu quá cao, chúng gây tổn thương nghiêm trọng gan, thận và hệ thần kinh. Cuối cùng, các triệu chứng bệnh tyrosine máu loại I khởi phát.
Chẩn đoán
Tyrosine máu loại I được chẩn đoán trước sinh bằng cách phát hiện succanylacetone và phân tích ADN của trẻ trong nước ối. Trong vòng 24–48 giờ sau khi trẻ chào đời, trẻ được lấy máu gót chân để thực hiện sàng lọc sơ sinh nhằm phát hiện tăng tyrosine máu loại I và các bệnh lý khác.
Nếu kết quả sàng lọc sơ sinh cho thấy nguy cơ cao mắc bệnh, bác sĩ sẽ cho em bé thực hiện thêm những xét nghiệm khác để làm rõ. Điều quan trọng cần lưu ý là kết quả sàng lọc nguy cơ cao không đồng nghĩa với kết luận em bé mắc bệnh. Kết quả nguy cơ cao có thể do mẫu máu ban đầu được thu quá ít hoặc quá sớm. Tuy nhiên, cha mẹ nên nhớ đưa trẻ tái khám theo đúng lịch hẹn để làm xét nghiệm xác nhận. Nếu không được điều trị kịp thời, tăng tyrosine máu loại I có thể ảnh hưởng sức khỏe của trẻ rõ rệt ngay sau khi sinh. Xét nghiệm tiếp theo phải được tiến hành càng sớm càng tốt để xác định xem liệu trẻ có mắc bệnh hay không.
Xét nghiệm tiếp theo bao gồm kiểm tra mẫu nước tiểu và máu của bé để đo lường nồng độ axit và chất độc có hại. Một số axit và độc tố tích tụ trong cơ thể khi trẻ bị rối loạn axit amin, do đó bác sĩ có thể dựa vào nồng độ các chất này để xác định xem em bé có mắc bệnh hay không. Trẻ mắc bệnh sẽ có hàm lượng chất chuyển hóa tyrosine và succanylacetone trong nước tiểu cao. Bên cạnh đó, bệnh có thể được chẩn đoán thông qua mức độ hoạt động của enzyme fumarylacetoacetate hydrolase trong mô gan hoặc nguyên bào sợi nuôi cấy. Ngoài ra, người bệnh có thể thực hiện xét nghiệm di truyền để xác nhận kết quả chẩn đoán.
Điều trị
Nếu được phát hiện sớm và điều trị kịp thời tyrosine máu loại I, trẻ có thể phòng ngừa các biến chứng nghiêm trọng cũng như duy trì sự phát triển, trí thông minh bình thường.
Phương pháp điều trị tăng tyrosine máu loại I cho bệnh nhân bao gồm:
- Sử dụng thuốc nitisinone nhằm ngăn ngừa tổn thương gan, thận và hệ thần kinh
- Duy trì chế độ ăn ít tyrosine/phenylalanine
- Bổ sung vitamin D và Carnitine trong một số trường hợp
Đối với mức độ nghiêm trọng như tại thời điểm chẩn đoán, người bệnh đã phát triển bệnh suy gan giai đoạn cuối, ung thư biểu mô tế bào gan hoặc không đáp ứng với thuốc nitisinone. Phương pháp điều trị cho những trường hợp này là phẫu thuật ghép gan. Phương pháp này cũng giúp người bệnh cải thiện chức năng thận.
Trẻ mắc bệnh tyrosine máu loại I cần xét nghiệm máu và nước tiểu thường xuyên để kiểm tra:
- Nồng độ axit amin
- Hàm lượng succanylaceton
- Mức độ nitisinone
- Chức năng gan và thận
Qua các xét nghiệm này, bác sĩ có thể theo dõi bệnh tiến triển nhằm điều chỉnh phác đồ điều trị, thay đổi thuốc hoặc chế độ ăn uống cho phù hợp nhất với tình trạng sức khỏe của trẻ. Ngoài ra, trẻ nên chụp CT hoặc MRI gan định kỳ mỗi năm một lần để kiểm tra sẹo gan hoặc ung thư gan.
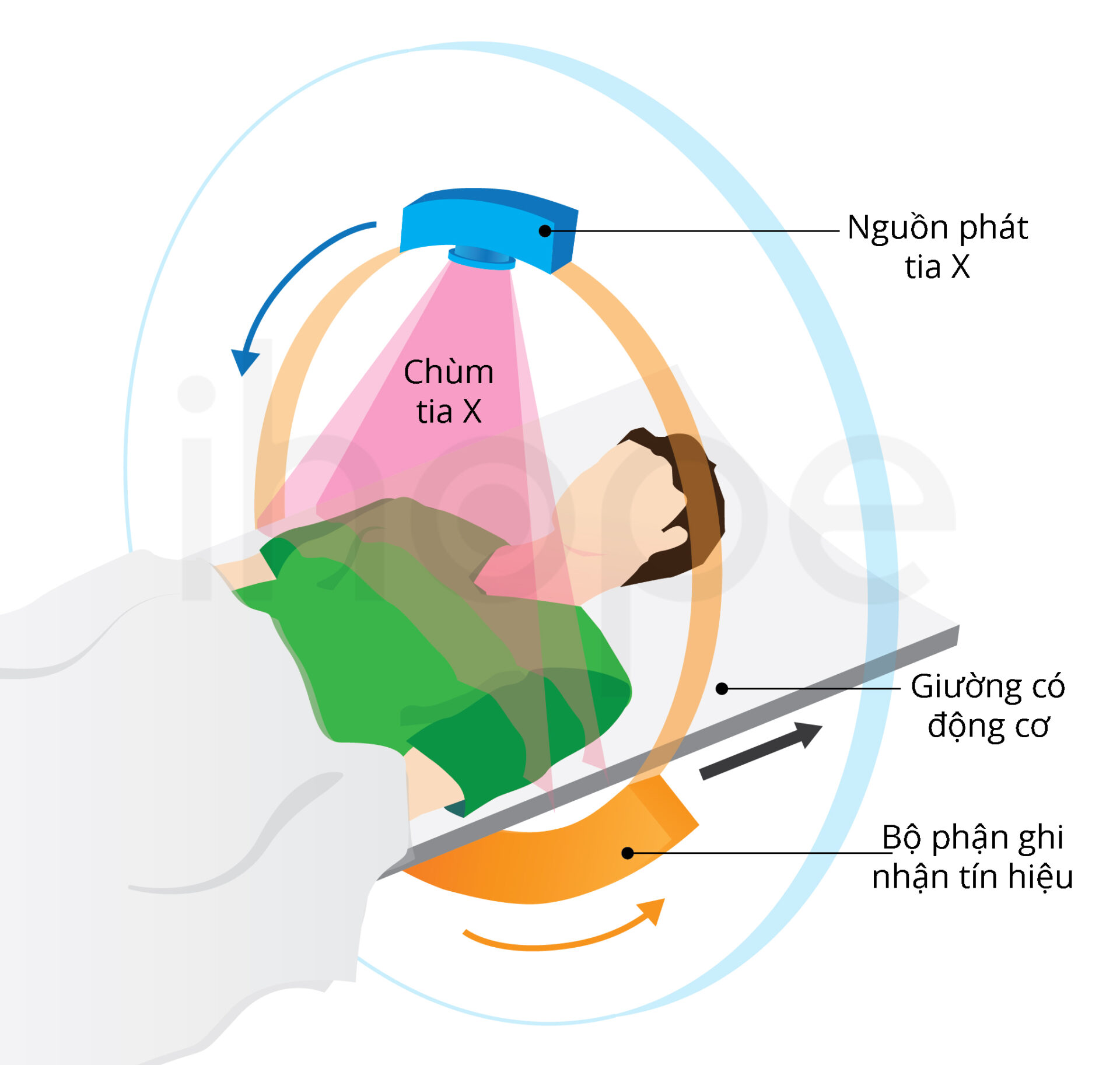
Nguồn: Lecturi
Dạng di truyền
Tăng tyrosine máu loại I di truyền theo kiểu lặn trên nhiễm sắc thể thường. Do đó, bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào. Bệnh nhân mắc bệnh lặn trên nhiễm sắc thể thường sẽ có bố và mẹ mang một bản sao của gen đột biến, nhưng họ ít khi biểu hiện triệu chứng bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Bệnh di truyền lặn do đột biến gen FAH, cha mẹ mang đột biến dị hợp nên gần như không có biểu hiện bệnh, do đó rất khó phát hiện cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn để chủ động cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kỳ.
Các tên gọi khác
- TYR I
- Tyrosinosis
- FAH deficiency
- Hypertyrosinemia
- Hereditary tyrosinemia
- Hereditary tyrosinemia type 1
- Heteditary Infantile tyrosinemia
- Hepatorenal tyrosinemia
- Fumarylacetoacetase deficiency
- Fumarylacetoacetate hydrolase deficiency
References
- Genetic Testing Information. Tyrosinemia type I. Retrieved January 02, 2024 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0268490/
- Genetic and Rare Diseases Information Center. Tyrosinemia type 1. Retrieved January 02, 2024 from https://rarediseases.info.nih.gov/diseases/2658/tyrosinemia-type-1
- Catalog of Genes and Diseases from OMIM. TYROSINEMIA, TYPE I; TYRSN1. Retrieved January 02, 2024 from https://www.omim.org/entry/276700#
- U.S National Library of Medicine. Tyrosinemia. Retrieved January 02, 2024 from https://medlineplus.gov/genetics/condition/tyrosinemia/
- Baby's First Test Tyrosinemia, Type I. Retrieved January 02, 2024 from https://www.babysfirsttest.org/newborn-screening/conditions/tyrosinemia-type-i
- Health Resources and Services Administration Tyrosinemia type I. Retrieved January 02, 2024 from https://newbornscreening.hrsa.gov/conditions/tyrosinemia-type-i
- National Institute of Health. Tyrosinemia Type I. Retrieved January 02, 2024 from https://www.ncbi.nlm.nih.gov/books/NBK1515/
- National Organization for Rare Disorders. Tyrosinemia Type 1. Retrieved January 02, 2024 from https://rarediseases.org/rare-diseases/tyrosinemia-type-1/
- Orphanet. Tyrosinemia type 1. Retrieved January 02, 2024 from https://www.orpha.net/consor4.01/www/cgi-bin/OC_Exp.php?lng=EN&Expert=882