
Hội chứng Feingold là bệnh di truyền ảnh hưởng đến nhiều bộ phận cơ thể. Bệnh bao gồm 2 dạng phân biệt dựa trên nguyên nhân di truyền. Mức độ nghiêm trọng các triệu chứng có thể khác nhau giữa từng bệnh nhân.
Biểu hiện lâm sàng
Hội chứng Fedingold loại 1 và 2 đều gây ra những bất thường ngón tay và ngón chân bao gồm:
- Ngón trỏ và ngón út ngắn (chứng brachymesophalangy)
- Tật lệch ngón (clinodactyly)
- Thiểu sản ngón cái
- Chứng dính khớp ngón tay

Ảnh: Ngón tay ngắn bất thường
Nguồn: U.S. National Library of Medicine
Ngoài ra, hội chứng còn khởi phát một số biểu hiện phổ biến như:
- Tật đầu nhỏ
- Cằm nhỏ
- Khe mí mắt hẹp
- Thiểu năng trí tuệ từ nhẹ đến trung bình
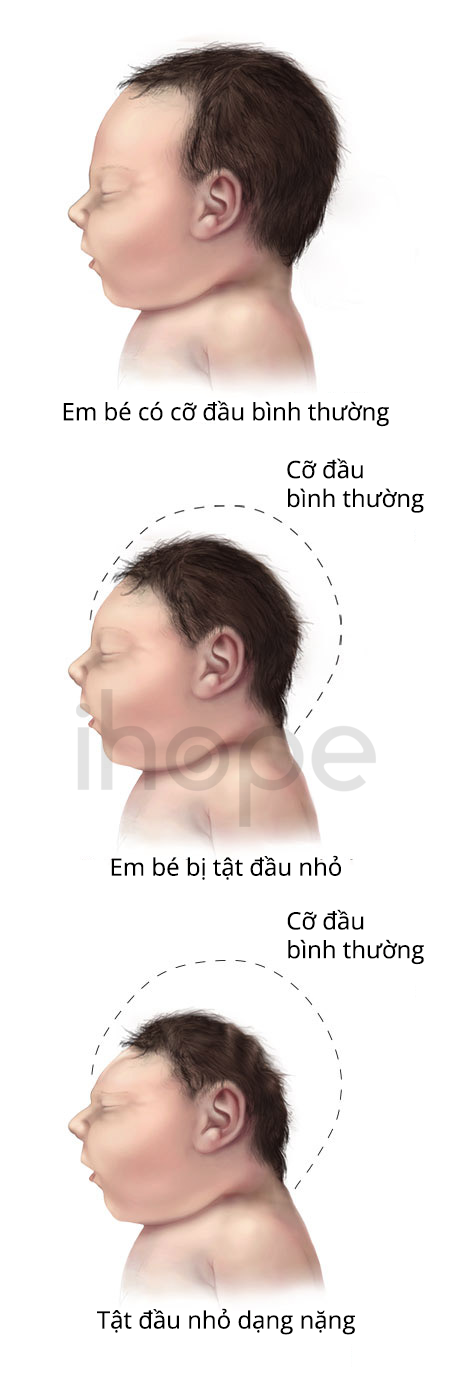
Ảnh: Tật đầu nhỏ
Nguồn: Centers for Disease Control and Prevention
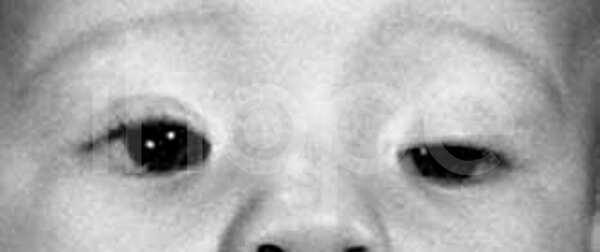
Ảnh: Khe mắt hẹp
Nguồn: National Human Genome Research Institute
Bệnh nhân có thể biểu hiện triệu chứng mất thính giác, tầm vóc thấp bé, bất thường thận và tim nhưng thường hiếm gặp.
Trẻ mắc hội chứng Feingold loại 1 thường bị hẹp đường tiêu hóa bẩm sinh. Phần lớn các trường hợp bị teo thực quản hoặc một phần tá tràng. Hội chứng loại 2 không gây ra các bất thường về đường tiêu hóa.
Độ phổ biến
Hội chứng Feingold là bệnh hiếm gặp, nên tỷ lệ mắc bệnh chưa được thống kê cụ thể. Người ta nhận thấy hội chứng loại 1 phổ biến hơn loại 2.
Nguyên nhân
Đột biến gen MYCN gây ra hội chứng Feingold loại 1. Đột biến mất đoạn bao gồm gen MIR17HG trên nhiễm sắc thể 13 gây ra hội chứng loại 2. Cả hai gen này đều liên quan đến quá trình tăng trưởng và phát triển chủ yếu trong giai đoạn trước sinh.
Gen MYCN cung cấp hướng dẫn tạo ra protein có chức năng điều hòa biểu hiện gen. Protein này liên kết với một số vị trí trên gen nhằm kiểm soát quá trình sản xuất protein. Người ta nhận thấy protein MYCN giúp tứ chi, tim, thận, phổi, hệ thần kinh và hệ tiêu hóa phát triển bình thường.
Gen MIR17HG cung cấp hướng dẫn tạo ra miR-17~92. Đây là những đoạn ARN ngắn giữ vai trò kiểm soát biểu hiện gen bằng cách ngăn chặn quá trình sản xuất protein. Nhiều mô và cơ quan thiết yếu cần miR-17~92 để phát triển.
Đột biến gen MYCN hoặc MIR17HG khiến protein MYCN và miR-17~92 mất chức năng. Do đó, quá trình phát triển của các cơ quan và tứ chi bị bất thường, từ đó các biểu hiện của hội chứng Feingold khởi phát.
Chẩn đoán
Hội chứng Feingold được chẩn đoán dựa trên những biểu hiện lâm sàng như tật đầu nhỏ, dị tật ngón tay, hẹp đường tiêu hóa bẩm sinh. Một số xét nghiệm hình ảnh như siêu âm, chụp cộng hưởng từ MRI có thể được chỉ định nhằm quan sát các bất thường. Ngoài ra, xét nghiệm di truyền phát hiện đột biến gen liên quan có thể được thực hiện.
Điều trị
Hiện nay chưa có phương pháp điều trị hoàn toàn hội chứng Feingold. Các liệu pháp chủ yếu giảm nhẹ triệu chứng nhằm cải thiện chất lượng đời sống bệnh nhân. Một số thủ thuật phẫu thuật có thể được chỉ định nhằm khắc phục chứng hẹp đường tiêu hóa bẩm sinh và dị tật ngón tay. Đối với chậm phát triển và thiểu năng trí tuệ, bệnh nhân cần hỗ trợ bằng chương trình giáo dục đặc biệt.
Dạng di truyền
Bệnh di truyền theo kiểu trội trên nhiễm sắc thể thường. Do đó, một bản sao của gen trong mỗi tế bào đủ để gây ra bệnh. Phần lớn người bệnh thừa hưởng một bản sao gen đột biến bệnh từ bố hoặc mẹ cũng mắc hội chứng.

Nguồn: U.S. National Library of Medicine
Ngoài ra, hội chứng có thể do đột biến mới (de novo) xảy ra trong quá trình tạo tế bào sinh sản (trứng và tinh trùng) hoặc phát triển phôi sớm. Những trường hợp này thường không có tiền sử bệnh trong gia đình.
Phòng ngừa
Bệnh di truyền trội trên nhiễm sắc thể thường, nếu cha hoặc mẹ mắc bệnh, con sinh ra sẽ có 50% khả năng di truyền bệnh. Do đó, để bảo đảm 100% khả năng con không bị bệnh, cha mẹ có thể chọn phương pháp thụ tinh nhân tạo IVF và sàng lọc phôi PGS/PGD. Các thành viên trong gia đình nên khám sức khỏe và tầm soát bệnh định kỳ nếu có thành viên mắc bệnh. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh.
Các tên gọi khác
- Brunner-Winter syndrome
- Microcephaly-mesobrachyphalangy-tracheoesophageal fistula (MMT) syndrome
- Microcephaly-oculo-digito-esophageal-duodenal (MODED) syndrome
- Oculo-digito-esophagoduodental (ODED) syndrome
References
- Genetic Testing Information. Feingold syndrome. Retrieved 31 July 2023 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0796068/?_ga=2.129352012.1188870066.1690769611-1635318420.1684225451
- Genetic and Rare Diseases Information Center. Feingold syndrome. Retrieved 31 July 2023 from https://rarediseases.info.nih.gov/diseases/8407/feingold-syndrome
- Catalog of Genes and Diseases from OMIM. FEINGOLD SYNDROME 1; FGLDS1. Retrieved 31 July 2023 from https://omim.org/entry/164280
- U.S National Library of Medicine. Feingold syndrome. Retrieved 31 July 2023 from https://medlineplus.gov/genetics/condition/feingold-syndrome/
- Frontiers. Occurrence of Esophageal Atresia With Tracheoesophageal Fistula in Siblings From Three-Generation Family Affected by Variable Expressivity MYCN Mutation: A Case Report. Retrieved 31 July 2023 from https://www.frontiersin.org/articles/10.3389/fped.2021.783553/full
- MalaCards. Feingold Syndrome 1 (FGLDS1). Retrieved 31 July 2023 from https://www.malacards.org/card/feingold_syndrome_1
- National Organization for Rare Disorders. Feingold syndrome. Retrieved 31 July 2023 from https://rarediseases.org/gard-rare-disease/feingold-syndrome/
- Orphanet. Feingold syndrome. Retrieved 31 July 2023 from https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=1305
- Radiopaedia. Feingold syndrome. Retrieved 31 July 2023 from https://radiopaedia.org/articles/feingold-syndrome





