
Hội chứng Holt-Oram (Holt-Oram syndrome) gây ra những bất thường chi trên và tim. Bệnh xảy ra trong giai đoạn thai kỳ nên ảnh hưởng lâu dài đến sức khỏe của trẻ. Các biến chứng nghiêm trọng có thể gây tử vong.
Biểu hiện lâm sàng
Người bệnh biểu hiện ít nhất một bất thường vùng xương cổ tay nhưng chỉ phát hiện bằng chụp X-quang.
- Thiếu ngón cái hoặc ngón cái dài bất thường
- Kém phát triển hoặc không có xương cánh tay
- Bất thường xương quai xanh, bả vai
- Dính xương ngón cái và cổ tay
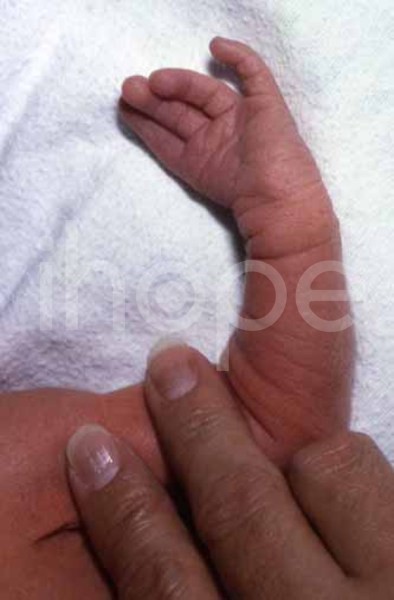
Ảnh: Thiếu ngón cái
Nguồn: U.S National Library of Medicine
Những bất thường này có thể xảy ra với một hoặc cả hai chi trên. Trường hợp cả hai chi trên bị ảnh hưởng, phần chi bên trái thường có triệu chứng nặng hơn bên phải.
Khoảng 75% người bệnh gặp vấn đề tim, phổ biến là dị tật vách ngăn tim gây ra thông liên nhĩ và thông liên thất. Trong thông liên nhĩ, một lỗ hỏng xuất hiện trên vách ngăn tâm nhĩ dẫn đến phần bên phải tim tăng khối lượng làm việc nên máu đến phổi nhiều hơn bình thường. Thông liên thất gây giảm hiệu quả bơm máu đến các cơ quan. Người bệnh có triệu chứng như thở nhanh, thở khò khè và nhịp tim nhanh. Nếu lỗ thông liên thất lớn có thể đe dọa tính mạng hoặc gây tổn thương động mạch phổi vĩnh viễn.
Các đặc điểm của hội chứng Holt-Oram tương tự như hội chứng Duane-radial ray nhưng khác nguyên nhân di truyền.
Độ phổ biến
Tỷ lệ mắc hội chứng Holt-Oram ước tính khoảng 1/100.000 người trên toàn thế giới.
Nguyên nhân
Khoảng 74% người mắc hội chứng Holt-Oram do đột biến gen TBX5 gây ra. Gen TBX5 cung cấp hướng dẫn tạo ra protein giữ vai trò quan trong quá trình hình thành vách ngăn tim và kiểm soát phát triển của xương các chi trên trong giai đoạn thai kỳ. Đột biến gen TBX5 dẫn đến phiên bản protein bất thường, ảnh hưởng đến quá trình phát triển rồi gây ra những triệu chứng của hội chứng Holt-Oram.
Những trường hợp còn lại chưa rõ nguyên nhân gây bệnh.
Chẩn đoán
Hội chứng Holt-Oram được chẩn đoán dựa trên bất thường thể chất, tiền sử bệnh của cá nhân và gia đình.
Một số xét nghiệm được chỉ định bao gồm:
- Chụp X-quang quan sát bất thường các chi trên
- Siêu âm tim và điện tâm đồ xác định các dị tật tim và mức độ nghiêm trọng
- Xét nghiệm di truyền tìm đột biến gen TBX5
Trường hợp có tiền sử bệnh tim trong gia đình, thai phụ nên siêu âm và thăm khám thai định kỳ.
Điều trị
Hiện nay chưa có phương pháp điều trị hội chứng Holt-Oram. Với phần lớn trường hợo, bác sĩ dựa vào triệu chứng cụ thể của mỗi người bệnh. Bất thường chi trên có thể cần phẫu thuật chỉnh hình hoặc tái tạo kèm theo vật lý trị liệu nhằm cải thiện khả năng vận động. Người bị dị tật tim có thể cần dùng thuốc, máy tạo nhịp tim nhân tạo hoặc phẫu thuật chỉnh sửa. Đồng thời, họ cần được siêu âm tim 1-5 năm/lần nhằm đánh giá quá trình điều trị hoặc ngăn ngừa các biến chứng nghiêm trọng.
Dạng di truyền
Bệnh di truyền theo kiểu trội trên nhiễm sắc thể thường. Một bản sao của gen đột biến trong mỗi tế bào đủ để gây ra bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Bệnh di truyền trội trên nhiễm sắc thể thường, nếu cha hoặc mẹ mắc bệnh, con sinh ra sẽ có 50% khả năng di truyền bệnh. Do đó, để bảo đảm 100% khả năng con không bị bệnh, cha mẹ có thể chọn phương pháp thụ tinh nhân tạo IVF và sàng lọc phôi PGS/PGD. Các thành viên trong gia đình nên khám sức khỏe và tầm soát bệnh định kỳ nếu có thành viên mắc bệnh. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh.
Các tên gọi khác
- Atrio-digital syndrome
- Atriodigital dysplasia
- Cardiac-limb syndrome
- Heart-hand syndrome, type 1
- HOS
- Ventriculo-radial syndrome
References
- Genetic Testing Information. Holt-Oram syndrome. Retrieved January 19, 2023 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0265264/
- Genetic and Rare Diseases Information Center. Holt-Oram syndrome. Retrieved January 19, 2023 from https://rarediseases.info.nih.gov/diseases/6666/holt-oram-syndrome
- Catalog of Genes and Diseases from OMIM. HOLT-ORAM SYNDROME. Retrieved January 19, 2023 from https://omim.org/entry/142900
- U.S National Library of Medicine. Holt-Oram syndrome. Retrieved January 19, 2023 from https://medlineplus.gov/genetics/condition/holt-oram-syndrome/
- National Organization for Rare Disorders. Holt-Oram syndrome. Retrieved January 19, 2023 from https://rarediseases.org/rare-diseases/holt-oram-syndrome/
- Orphanet. Holt-Oram syndrome. Retrieved January 19, 2023 from https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=GB&Expert=392
- Basson CT, Cowley GS, Solomon SD, Weissman B, Poznanski AK, Traill TA, Seidman JG, Seidman CE. The clinical and genetic spectrum of the Holt-Oram syndrome (heart-hand syndrome). N Engl J Med. 1994 Mar 31;330(13):885-91. doi: 10.1056/NEJM199403313301302
- Cerbai E, Sartiani L. Holt-oram syndrome and atrial fibrillation: opening the (T)-box. Circ Res. 2008 Jun 6;102(11):1304-6. doi: 10.1161/CIRCRESAHA.108.178079
- Boogerd CJ, Dooijes D, Ilgun A, Mathijssen IB, Hordijk R, van de Laar IM, Rump P, Veenstra-Knol HE, Moorman AF, Barnett P, Postma AV. Functional analysis of novel TBX5 T-box mutations associated with Holt-Oram syndrome. Cardiovasc Res. 2010 Oct 1;88(1):130-9. doi: 10.1093/cvr/cvq178
- Chryssostomidis G, Kanakis M, Fotiadou V, Laskari C, Kousi T, Apostolidis C, Azariadis P, Chatzis A. Diversity of congenital cardiac defects and skeletal deformities associated with the Holt-Oram syndrome. Int J Surg Case Rep. 2014;5(7):389-92. doi: 10.1016/j.ijscr.2014.04.034





