Hội chứng mất đoạn 17q12 xảy ra do một đoạn nhỏ bị xóa trên nhiễm sắc thể số 17, người bệnh gặp các vấn đề nghiêm trọng về thận và hệ tiết niệu. Người bệnh có thể phát triển bệnh tiểu đường sớm kèm theo những bất thường thần kinh.
Biểu hiện lâm sàng
Các dấu hiệu và triệu chứng của hội chứng mất đoạn 17q12 biểu hiện khác nhau, ngay cả giữa các thành viên trong một gia đình.
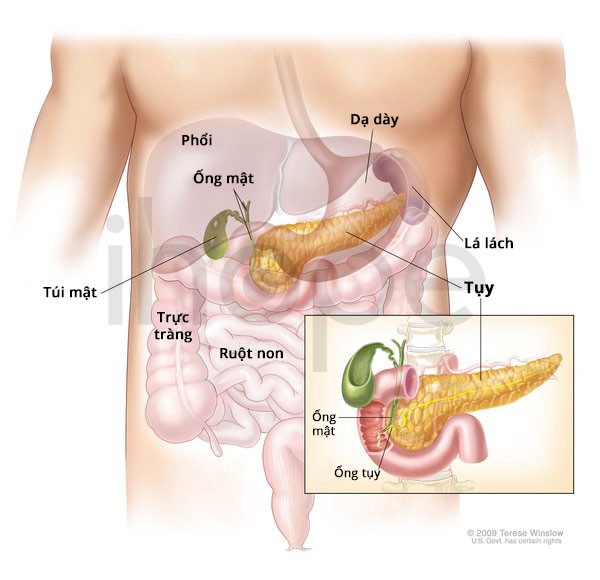
Người bệnh thường gặp vấn đề phát triển hoặc chức năng của thận và hệ tiết niệu. Trẻ bị suy thận trước khi sinh. Nhiều trường hợp phát triển bệnh tiểu đường trước 25 tuổi (Maturity-Onset Diabetes of the Young type 5 - MODY5). Bệnh xảy ra do một số tế bào bất thường trong tuyến tụy.
Nguồn: Terese Winslow LLC
Khoảng 50% trẻ mắc bệnh chậm phát triển các kỹ năng như chậm nói, chậm phát triển ngôn ngữ.
Các biểu hiện liên quan đến thần kinh bao gồm:
- Thiểu năng trí tuệ
- Tự kỷ
- Tâm thần phân liệt
- Rối loạn lưỡng cực
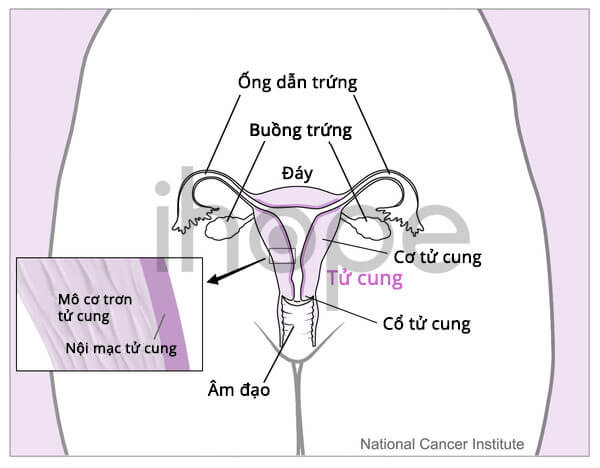
Trường hợp hiếm gặp, bệnh gây ra những bất thường tại mắt, gan, não và cơ quan sinh dục. Nữ giới mắc bệnh phát triển hội chứng Mayer-Rokitansky-Küster-Hauser. Họ thường kém phát triển, không có âm đạo và tử cung. Một số trẻ có khuôn mặt dị biệt.
Nguồn: NIH Medical Arts/National Cancer Institute
Độ phổ biến
Hội chứng mất đoạn 17q12 hiếm gặp, do đó hiện nay tỉ lệ mắc bệnh trên toàn thế giới vẫn chưa được thống kê cụ thể. Riêng tại Iceland, ước tính có khoảng 1/14.500 người bệnh.
Nguyên nhân
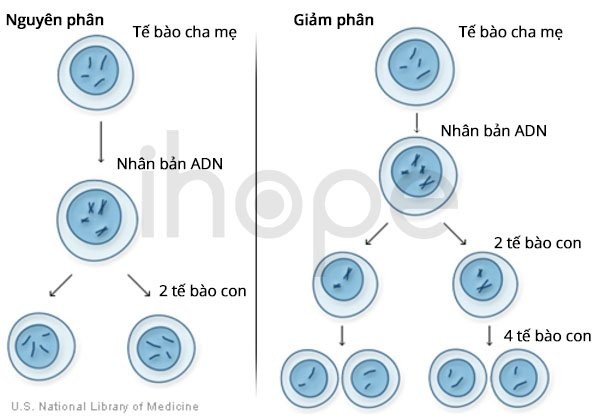
Phần lớn người mắc hội chứng mất đoạn 17q12 thiếu khoảng 1,4 triệu cặp base (1,4 Mb) tại vị trí q12 trên nhiễm sắc thể số 17. Hiện tượng này ảnh hưởng một trong hai bản sao của nhiễm sắc thể 17 trong mỗi tế bào. Đoạn bị xóa được bao quanh bởi các chuỗi ADN ngắn, lặp đi lặp lại khiến cho đoạn này dễ bị sắp xếp lại trong quá trình phân chia tế bào. Quá trình sắp xếp lại có thể dẫn đến thiếu hoặc thừa các bản sao ADN tại 17q12.
Nguồn: U.S. National Library of Medicine
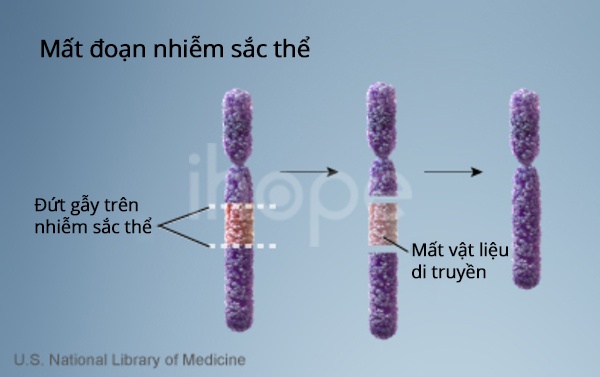
Ảnh: Đột biến mất đoạn nhiễm sắc thể
Nguồn: U.S. National Library of Medicine
Phần lớn người bệnh thường mất khoảng 15 gen. Mất hai gen HNF1B và LHX1 là cơ sở gây ra một số đặc điểm của bệnh. Theo các nghiên cứu, mất một bản sao của gen HNF1B trong mỗi tế bào dẫn đến bất thường về thận, đường tiết niệu và tuyến tụy. Mất một bản sao LHX1 góp phần gây ra thiểu năng trí tuệ, rối loạn hành vi và tâm thần, hội chứng Mayer-Rokitansky-Küster-Hauser. Thiếu các gen khác trong vùng bị xóa làm tăng nguy cơ xuất hiện các triệu chứng hiếm gặp của hội chứng mất đoạn 17q12.
Chẩn đoán
Bệnh được chẩn đoán bệnh dựa vào biểu hiện lâm sàng, tiền sử bệnh của cá nhân và gia đình. Bác sĩ sẽ chỉ định một số xét nghiệm chuyên sâu hỗ trợ chẩn đoán và phân biệt với các bệnh lí khác.
Một số xét nghiệm có thể thực hiện bao gồm:
- Xét nghiệm hình ảnh (chụp CT, siêu âm) quan sát các bất thường về thận, đường tiết niệu
- Xét nghiệm sinh hóa nhằm đánh giá chức năng thận
- Các bài kiểm tra, chẩn đoán về tâm thần
- Xét nghiệm di truyền xác định đột biến mất gen liên quan
Điều trị
Hiện nay chưa có phương pháp điều trị hoàn toàn hội chứng mất đoạn 17q12. Các liệu pháp chủ yếu giảm nhẹ triệu chứng của mỗi người bệnh nhằm cải thiện chất lượng cuộc sống của họ.
Các liệu pháp được áp dụng bao gồm:
- Suy thận, dị tật thận có thể được lọc máu hoặc ghép thận
- Thiểu năng trí tuệ, tâm thần phân liệt cần phối hợp giữa giáo dục và chăm sóc đặc biệt
- Điều trị tiểu đường bằng thuốc hạ đường huyết hoặc insulin theo chỉ định của bác sĩ
- Bất thường cơ quan sinh dục có thể can thiệp phẫu thuật điều chỉnh
Dạng di truyền
Phần lớn các trường hợp mắc hội chứng mất đoạn 17q12 do đột biến mới (de novo) trong quá trình sản sinh tế bào sinh sản (trứng và tinh trùng) hoặc tạo phôi. Những bệnh nhân này không có tiền sử mắc hội chứng trong gia đình của họ.
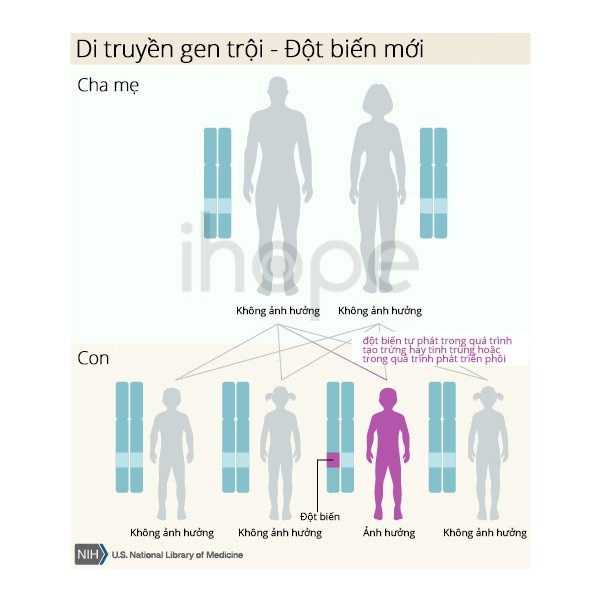
Nguồn: U.S. National Library of Medicine
Trường hợp hiếm gặp bệnh di truyền theo kiểu trội trên nhiễm sắc thể thường. Tế bào mang một bản sao nhiễm sắc thể bị mất đoạn đủ để gây ra bệnh. Con cái có thể thừa hưởng nhiễm sắc thể bị mất đoạn từ cha hoặc mẹ bị bệnh.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Hội chứng mất đoạn 17q12 di truyền trội trên nhiễm sắc thể thường, nếu cha hoặc mẹ mắc bệnh, con sinh ra sẽ có 50% khả năng di truyền bệnh. Do đó, để bảo đảm 100% khả năng con không bị bệnh, cha mẹ có thể chọn phương pháp thụ tinh nhân tạo IVF và sàng lọc phôi PGS/PGD.
Các thành viên trong gia đình nên khám sức khỏe và tầm soát bệnh định kỳ nếu có thành viên mắc bệnh. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh.
Các tên gọi khác
- 17q12 chromosomal microdeletion
- 17q12 microdeletion
- 17q12 recurrent deletion syndrome
- Deletion 17q12
- Recurrent genomic rearrangement in chromosome 17q12
References
- Genetic Testing Information. Chromosome 17q12 deletion syndrome. Retrieved December 23, 2022 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C3281138/
- Catalog of Genes and Diseases from OMIM. CHROMOSOME 17q12 DELETION SYNDROME. Retrieved December 23, 2022 from https://omim.org/entry/614527
- U.S National Library of Medicine. 17q12 deletion syndrome. Retrieved December 23, 2022 from https://medlineplus.gov/genetics/condition/17q12-deletion-syndrome/
- Orphanet. 17q12 microdeletion syndrome. Retrieved December 23, 2022 from https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=EN&Expert=261265
- MalaCards. Chromosome 17q12 Deletion Syndrome. Retrieved December 23, 2022 from https://www.malacards.org/card/chromosome_17q12_deletion_syndrome
- National Organization for Rare Disorders. 17q12 deletion syndrome. Retrieved December 23, 2022 from https://rarediseases.org/gard-rare-disease/17q12-deletion-syndrome/
- Laffargue F, Bourthoumieu S, Llanas B, Baudouin V, Lahoche A, Morin D, Bessenay L, De Parscau L, Cloarec S, Delrue MA, Taupiac E, Dizier E, Laroche C, Bahans C, Yardin C, Lacombe D, Guigonis V. Towards a new point of view on the phenotype of patients with a 17q12 microdeletion syndrome. Arch Dis Child. 2015 Mar;100(3):259-64. doi: 10.1136/archdischild-2014-306810.
- Mitchel MW, Moreno-De-Luca D, Myers SM, Levy RV, Turner S, Ledbetter DH, Martin CL. 17q12 Recurrent Deletion Syndrome. 2016 Dec 8 [updated 2020 Oct 15]. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2022. Available from http://www.ncbi.nlm.nih.gov/books/NBK401562/
- Nagamani SC, Erez A, Shen J, Li C, Roeder E, Cox S, Karaviti L, Pearson M, Kang SH, Sahoo T, Lalani SR, Stankiewicz P, Sutton VR, Cheung SW. Clinical spectrum associated with recurrent genomic rearrangements in chromosome 17q12. Eur J Hum Genet. 2010 Mar;18(3):278-84. doi: 10.1038/ejhg.2009.174.
- Rasmussen M, Vestergaard EM, Graakjaer J, Petkov Y, Bache I, Fagerberg C, Kibaek M, Svaneby D, Petersen OB, Brasch-Andersen C, Sunde L. 17q12 deletion and duplication syndrome in Denmark-A clinical cohort of 38 patients and review of the literature. Am J Med Genet A. 2016 Nov;170(11):2934-2942. doi: 10.1002/ajmg.a.37848.