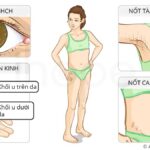
U cận hạch không hội chứng (nonsyndromic paraganglioma) là một bệnh lí với đặc điểm nổi bật là sự phát triển của các khối u trong hạch thần kinh (paraganglia). Hạch thần kinh bao gồm nhiều nhóm tế bào thuộc hệ thần kinh ngoại biên phân tán rộng rãi trong cơ thể. Các khối u thường xuất hiện nhiều nhất tại vùng đầu, cổ, ngực và bụng. Điểm khác biệt quan trọng của bệnh này so với các hội chứng di truyền gây u hạch thần kinh khác là bệnh nhân chỉ có các khối u đơn lẻ, không kèm theo bất kì triệu chứng bất thường nào tại các cơ quan khác hay các biểu hiện của một hội chứng toàn thân.
Biểu hiện lâm sàng
Các triệu chứng của u cận hạch không hội chứng phụ thuộc vào vị trí và kích thước của khối u. Đa số các khối u xuất hiện tại vùng đầu và cổ, chủ yếu xung quanh động mạch cảnh (carotid body)—cơ quan cảm nhận nồng độ oxy trong máu. Những khối u này thường phát triển chậm và không tiết ra hormone, nhưng chúng có khả năng chèn ép các cấu trúc lân cận như dây thần kinh sọ, do đó bệnh nhân có các triệu chứng như mất thính lực, ù tai, khó nuốt hoặc khàn tiếng.
Một số khối u hạch thần kinh khác tại vùng bụng hoặc ngực có khả năng sản xuất quá mức các hormone catecholamine (như adrenaline hoặc noradrenaline). Tình trạng gia tăng nồng độ catecholamine trong máu gây ra các cơn tăng huyết áp kịch phát, nhịp tim nhanh, vã mồ hôi và đau đầu dữ dội. Trong một số trường hợp, khối u có thể trở thành ác tính và di căn đến các hạch bạch huyết, xương, phổi hoặc gan.
Độ phổ biến
Người ta ước tính tỉ lệ mắc u cận hạch không hội chứng vào khoảng 1/1.000.000 đến 1/500.000 người. Bệnh chiếm khoảng 10% đến 20% tổng số các trường hợp u hạch thần kinh. Tuy nhiên, rất khó xác định được tỉ lệ chính xác do nhiều trường hợp khối u phát triển chậm và không gây ra triệu chứng lâm sàng rõ rệt trong thời gian dài.
Nguyên nhân
U cận hạch không hội chứng bắt nguồn từ đột biến tại các gen thuộc phức hợp enzyme succinate dehydrogenase (SDH), bao gồm SDHA, SDHB, SDHC và SDHD. Phức hợp enzyme này nắm vai trò trọng yếu trong ti thể, nó tham gia vào chu trình Krebs và chuỗi truyền electron để tạo năng lượng cho tế bào. Đột biến các gen SDH khiến tế bào giảm khả năng kiểm soát sự phát triển và các chất chuyển hóa trung gian tích tụ, từ đó các đường tín hiệu được kích hoạt để khối u dần hình thành. Ngoài ra, đột biến tại gen SDHAF2 hoặc MAX cũng được xác định là nguyên nhân gây bệnh trong một số ít trường hợp.
Chẩn đoán
Bệnh nhân được chẩn đoán u cận hạch không hội chứng dựa trên triệu chứng lâm sàng, xét nghiệm sinh hóa và chẩn đoán hình ảnh. Xét nghiệm định lượng catecholamine và các chất chuyển hóa metanephrine trong nước tiểu hoặc huyết tương có thể cho thấy hoạt động nội tiết của khối u. Các kĩ thuật chẩn đoán hình ảnh như chụp cắt lớp vi tính (CT) hoặc chụp cộng hưởng từ (MRI) được sử dụng để xác định vị trí, kích thước và sự xâm lấn của khối u. Chụp xạ hình chức năng (PET/CT) với các chất đánh dấu đặc hiệu có thể phát hiện các ổ di căn hoặc các khối u nhỏ phân tán khắp cơ thể. Xét nghiệm di truyền nắm vai trò quyết định để xác định đột biến gen, từ đó bác sĩ có thể phân loại nguy cơ và tư vấn cho các thành viên trong gia đình.
Điều trị
Phương pháp điều trị chủ yếu đối với u cận hạch không hội chứng là phẫu thuật cắt bỏ khối u. Đối với các khối u vùng đầu và cổ không có khả năng phẫu thuật hoặc khối u nhỏ không gây triệu chứng, liệu pháp xạ trị hoặc theo dõi định kì có thể được cân nhắc. Trong trường hợp khối u bài tiết catecholamine, bệnh nhân cần được điều trị bằng thuốc chẹn alpha và beta-adrenergic trước khi can thiệp ngoại khoa nhằm kiểm soát huyết áp và ngăn ngừa các biến chứng tim mạch trong quá trình phẫu thuật. Đối với các trường hợp ác tính đã di căn, các liệu pháp bổ trợ như hóa trị, xạ trị đích bằng dược chất phóng xạ (như MIBG hoặc liệu pháp thụ thể peptide) được áp dụng nhằm kiểm soát sự phát triển của khối u và giảm nhẹ triệu chứng.
Dạng di truyền
U cận hạch không hội chứng di truyền theo kiểu trội trên nhiễm sắc thể thường, nghĩa là chỉ cần một bản sao của gen đột biến trong mỗi tế bào đã đủ để tăng nguy cơ hình thành khối u. Tuy nhiên, bệnh thể hiện đặc điểm in vết di truyền (genomic imprinting) đối với các đột biến gen SDHD và SDHAF2. Do đó, nguy cơ mắc bệnh chỉ biểu hiện khi gen đột biến được di truyền từ người cha. Cá nhân nhận gen đột biến từ người mẹ thường không phát triển khối u nhưng vẫn có khả năng truyền gen này cho thế hệ sau.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
U cận hạch không hội chứng mang tính chất di truyền trội, người mang đột biến gen có nguy cơ cao phát triển khối u trong suốt cuộc đời. Để chủ động phòng ngừa, các thành viên trong gia đình bệnh nhân nên thực hiện xét nghiệm di truyền nhằm xác định tình trạng mang gen đột biến. Những cá nhân có kết quả dương tính cần thực hiện tầm soát định kì bằng chẩn đoán hình ảnh và xét nghiệm sinh hóa nhằm phát hiện sớm các khối u khi chúng còn nhỏ và chưa gây biến chứng. Các cặp vợ chồng có tiền sử gia đình mắc bệnh nên thực hiện tư vấn di truyền và các xét nghiệm chẩn đoán trước sinh hoặc sàng lọc phôi nhằm đảm bảo khả năng sinh con khỏe mạnh.
Các tên gọi khác
- Familial paraganglioma
- Glomus tumor
- Hereditary paraganglioma-pheochromocytoma syndrome
- Paraganglioma, familial nonchromaffin
References
- National Library of Medicine. Pheochromocytoma. Retrieved February 15, 2026 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C0031511/
- NORD. Paragangliomas 1. Retrieved February 15, 2026 from https://rarediseases.info.nih.gov/diseases/7324/index
- OMIM. PHEOCHROMOCYTOMA. Retrieved February 15, 2026 from https://www.omim.org/entry/171300
- MedlinePlus. Nonsyndromic paraganglioma. Retrieved February 15, 2026 from https://medlineplus.gov/genetics/condition/nonsyndromic-paraganglioma/
- StoryMD. Diagnosis: Nonsyndromic Paraganglioma. Retrieved February 15, 2026 from https://storymd.com/journal/5mrq3pn0zw-nonsyndromic-paraganglioma/page/x9la8s2ge5-diagnosis
- StoryMD. Treatment: Nonsyndromic Paraganglioma. Retrieved February 15, 2026 from https://storymd.com/journal/5mrq3pn0zw-nonsyndromic-paraganglioma/page/zkon9trkqd-treatment