
Tăng sản thượng thận nguyên phát (primary macronodular adrenal hyperplasia) là bệnh di truyền hiếm gặp, nguyên nhân chủ yếu bắt nguồn từ đột biến gen ARMC5 trên nhiễm sắc thể 16. Biểu hiện lâm sàng của bệnh bao gồm nhiều khối u tại tuyến thượng thận, tăng nồng độ hormone cortisol trong máu, loãng xương và mệt mỏi kéo dài.
Biểu hiện lâm sàng
Tuyến thượng thận là cặp tuyến nhỏ nằm phía trên mỗi quả thận, chúng sản xuất các hormone thiết yếu như cortisol, aldosterone, adrenaline và noradrenaline. Bệnh tăng sản thượng thận nguyên phát xảy ra khi các khối u lớn hơn 1 cm hình thành bên trong tuyến thượng thận, dẫn đến tuyến này phát triển lớn bất thường và tăng sản xuất hormone cortisol.
Nồng độ cortisol trong máu cao hơn bình thường dẫn đến các triệu chứng như:
- Tăng cân, biểu hiện rõ tại mặt và thân trên
- Da mỏng, dễ bầm tím
- Loãng xương
- Yếu cơ
- Mệt mỏi
- Tăng huyết áp
- Tiểu đường
Bệnh thường khởi phát vào độ tuổi 40–50 và có thể tiến triển thành hội chứng Cushing nội sinh khi nhiều cortisol tích tụ trong cơ thể.
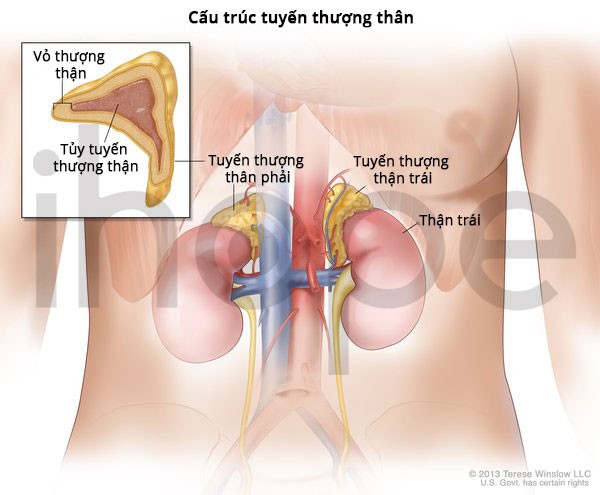
Nguồn: U.S. National Library of Medicine
Độ phổ biến
Bệnh tăng sản thượng thận nguyên phát rất hiếm gặp. Hiện nay, khoảng 170 trường hợp mắc bệnh đã được ghi nhận.
Nguyên nhân
Hơn một nửa số trường hợp mắc bệnh tăng sản thượng thận nguyên phát bắt nguồn từ đột biến gen ARMC5 trên nhiễm sắc thể 16. Gen ARMC5 cung cấp hướng dẫn tạo ra protein có chức năng ức chế khối u. Protein này tham gia cơ chế ngăn ngừa tế bào phát triển và phân chia quá mức. Đột biến gen ARCM5 khiến protein ức chế hình thành khối u không hoạt động, dẫn đến các tế bào tuyến thượng thận tăng sinh mất kiểm soát rồi tạo thành khối u.
Ngoài ra, người ta cũng ghi nhận những trường hợp mắc bệnh liên quan đến một số gen như KDM1A/LSD1, GNAS, PDE11A, PRKACA và MC2R. Phần lớn các gen này nắm vai trò trọng yếu trong đường tín hiệu sản xuất cortisol. Do đó, đột biến gen khiến tuyến thượng thận hoạt động sai nên quá nhiều cortisol được sản xuất rồi cuối cùng những triệu chứng bệnh khởi phát.
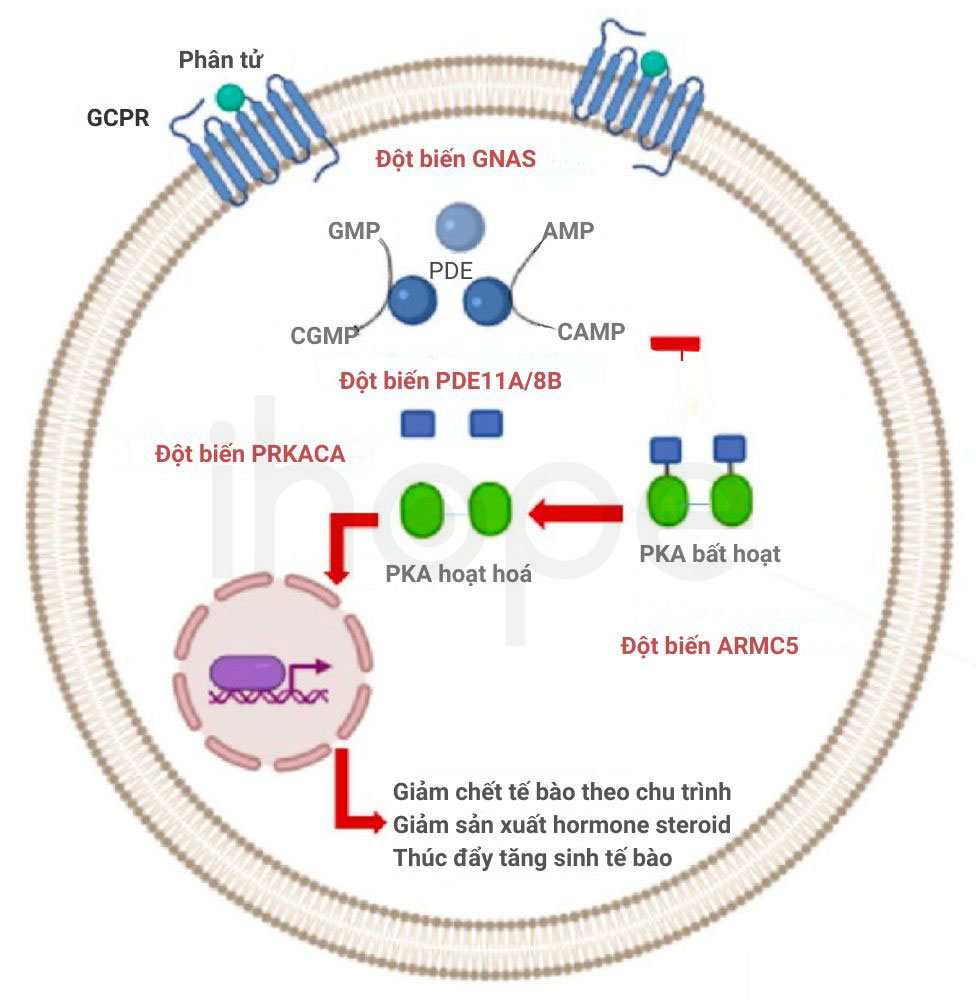
Nguồn: Diagnostics
Chẩn đoán
Tăng sản thượng thận nguyên phát được chẩn đoán dựa trên biểu hiện lâm sàng, bệnh sử gia đình kết hợp với kết quả xét nghiệm sinh hóa, hình ảnh và di truyền.
Xét nghiệm sinh hoá
Bác sĩ kiểm tra nồng độ cortisol trong máu và nước tiểu của bệnh nhân nhằm đưa ra chẩn đoán ban đầu. Đồng thời, bệnh nhân được chỉ định dùng thuốc dexamethasone để đánh giá khả năng ức chế sản xuất cortisol của tuyến yên—cơ quan điều hòa hoạt động của tuyến thượng thận. Nồng độ cortisol trong máu không giảm sau khi dùng thuốc cho thấy cơ thể mất khả năng điều hòa hormone. Ngoài ra, bác sĩ có thể chỉ định thêm các xét nghiệm chuyên sâu khác nhằm loại trừ những bệnh lí có biểu hiện tương tự.
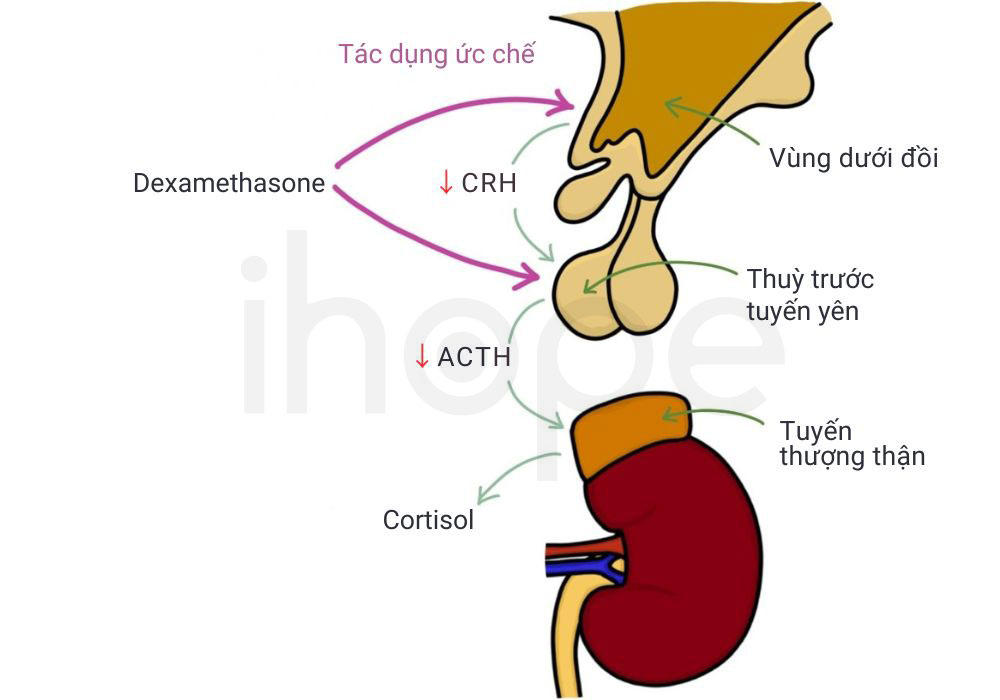
Nguồn: Zero to Finals
Chẩn đoán hình ảnh
Các phương pháp chẩn đoán hình ảnh như chụp cộng hưởng từ (MRI), chụp cắt lớp phát xạ positron (PET) hoặc chụp cắt lớp (CT) giúp bác sĩ phát hiện những biểu hiện bất thường trong tuyến thượng thận của bệnh nhân.

Nguồn: Cleverland Clinic
Xét nghiệm di truyền
Bác sĩ thực hiện các xét nghiệm di truyền nhằm phát hiện đột biến gen gây bệnh, qua đó kết quả chẩn đoán được xác nhận.
Điều trị
Phương pháp điều trị chính đối với bệnh tăng sản thượng thận nguyên phát là phẫu thuật cắt bỏ tuyến thượng thận. Bác sĩ có thể chỉ định cắt bỏ một phần hoặc toàn bộ tuyến tùy vào mức độ hoạt động của mỗi tuyến. Trong trường hợp bệnh nhân không thể phẫu thuật hoặc bác sĩ cần kiểm soát một số triệu chứng trước mổ, bệnh nhân có thể được điều trị bằng thuốc ức chế tổng hợp cortisol như ketoconazole, metyrapone, mitotane, trilostane và mifepristone. Sau phẫu thuật, bệnh nhân cần dùng thuốc thay thế hormone suốt đời nhằm phòng ngừa suy tuyến thượng thận.
Dạng di truyền
Tăng sản thượng thận nguyên phát chủ yếu di truyền theo kiểu trội trên nhiễm sắc thể thường. Do đó, một bản sao của gen đột biến trong mỗi tế bào đủ để gây ra bệnh. Người bệnh thường được di truyền từ cha hoặc mẹ mắc bệnh. Một số ít trường hợp là do đột biến mới (de novo) xuất hiện trong quá trình hình thành tế bào sinh sản (trứng, tinh trùng) hoặc quá trình phát triển phôi sớm. Ngoài ra, người ta cũng ghi nhận các trường hợp di truyền theo kiểu lặn trên nhiễm sắc thể thường—bệnh chỉ biểu hiện khi có cả hai bản sao của gen đột biến trong mỗi tế bào.

Nguồn: U.S. National Library of Medicine
Phòng ngừa
Đối với trường hợp di truyền trội trên nhiễm sắc thể thường, nếu cha hoặc mẹ mắc bệnh, con sinh ra sẽ có 50% khả năng di truyền bệnh. Đối với trường hợp di truyền theo kiểu lặn trên nhiễm sắc thể thường, bệnh thường khó được phát hiện trên các cặp cha mẹ mang gen bệnh cho đến khi sinh con. Để chủ động phòng ngừa, cha mẹ nên làm xét nghiệm sàng lọc gen lặn cho tương lai của con. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền đảm bảo sinh con khỏe mạnh. Người thuộc nhóm nguy cơ mắc bệnh cần khám sức khỏe và tầm soát bệnh định kì.
Các tên gọi khác
- ACTH-independent macronodular adrenal hyperplasia
- ACTH-independent macronodular adrenocortical hyperplasia
- Adrenal Cushing syndrome due to AIMAH
- Adrenocorticotropic hormone-independent macronodular adrenal hyperplasia
- AIMAH
- Corticotropin-independent macronodular adrenal hyperplasia
- PMAH
- Primary bilateral macronodular adrenal hyperplasia
- PBMAH
- Nodular cortical adrenal hyperplasia
- Massive macronodular hyperplasia
- Giant macronodular adrenal hyperplasia
References
- MedlinePlus. Primary macronodular adrenal hyperplasia. Retrieved August 7, 2025 from https://medlineplus.gov/genetics/condition/primary-macronodular-adrenal-hyperplasia/
- Oxford Academic. An Overview of the Heterogeneous Causes of Cushing Syndrome Resulting From Primary Macronodular Adrenal Hyperplasia (PMAH). Retrieved August 7, 2025 from https://doi.org/10.1210/jendso/bvac041
- Springer Nature. Primary bilateral macronodular adrenocortical hyperplasia (PBMAH) patient with ARMC5 mutations. Retrieved August 7, 2025 from https://doi.org/10.1186/s12902-023-01324-3
- Nature. Primary bilateral macronodular adrenal hyperplasia: definitely a genetic disease. Retrieved August 7, 2025 from https://www.nature.com/articles/s41574-022-00718-y
- Oxford Academic. Clinical, Pathophysiologic, Genetic, and Therapeutic Progress in Primary Bilateral Macronodular Adrenal Hyperplasia. Retrieved August 7, 2025 from https://doi.org/10.1210/endrev/bnac034





