Hội chứng khuynh hướng khối u cơ vân (rhabdoid tumor predisposition syndrome) khiến người bệnh có nguy cơ cao hình thành khối u cơ vân ác tính. Tế bào khối u có hình dạng giống với nguyên bào cơ vân (nguyên bào cơ xương), chúng có khả năng xâm lấn mạnh đến những mô xung quanh. Nguyên bào cơ vân là những tế bào hiện diện trong phôi thai, trong quá trình phát triển phôi thai, chúng hợp nhất với nhau để hình thành nên cơ xương.
Nguồn: U.S. National Library of Medicine

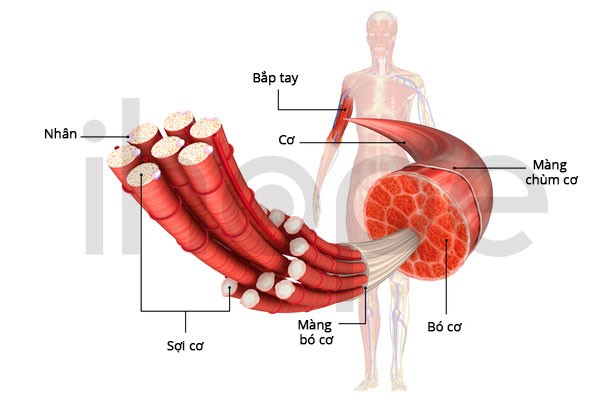
Ảnh: Các loại mô cơ
Nguồn: U.S. National Library of Medicine
Biểu hiện lâm sàng
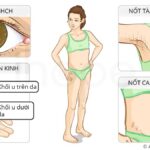
Hội chứng khuynh hướng u cơ vân thường xuất hiện trong những năm đầu sau sinh, trẻ ít có khả năng mắc bệnh sau 4 tuổi. Độ tuổi trung bình khởi phát bệnh từ 4–7 tháng và có thể xảy ra trước sinh. Khối u cơ vân xuất hiện tại nhiều vị trí khác nhau trong cơ thể (khối u đa ổ nguyên phát), chúng có xu hướng phát triển và lan rộng rất nhanh so với các dạng khối u khác. Do đó, nhiều bệnh nhân thường tử vong trong giai đoạn trẻ nhỏ.
Phần lớn khối u cơ vân ác tính hình thành tại tiểu não—vùng não điều phối vận động. Người ta gọi những khối u này là u quái không điển hình.
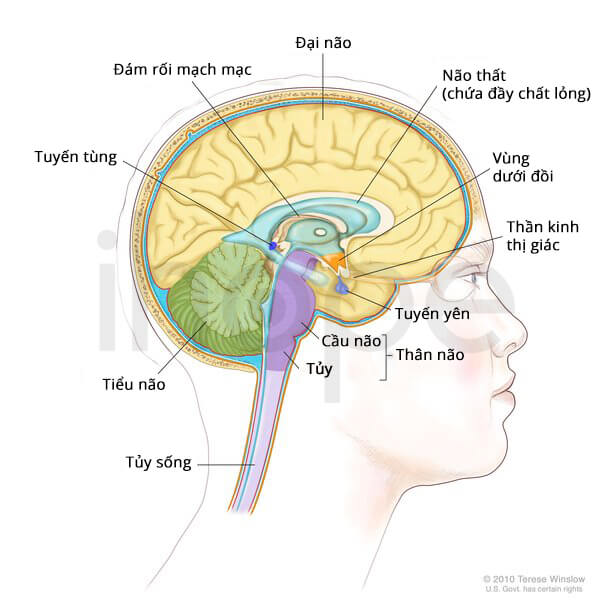
Nguồn: Terese Winslow
Ngoài hệ thần kinh trung ương, khối u cơ vân có thể xuất hiện tại nhiều vị trí khác trong cơ thể như khối u cơ vân thận và khối u cơ vân ác tính ngoài thận. Chúng có hình dạng, kích thước và vị trí khác nhau.
Nguồn: National Cancer Institue
Bên cạnh khối u cơ vân, người bệnh cũng có nguy cơ hình thành các khối u khác. Một số trẻ em mắc bệnh có thể phát triển khối u lành tính schwannoma, chúng phát triển từ tế bào Schwann—tế bào bao quanh và bảo vệ các sợi thần kinh trong hệ thần kinh ngoại vi.
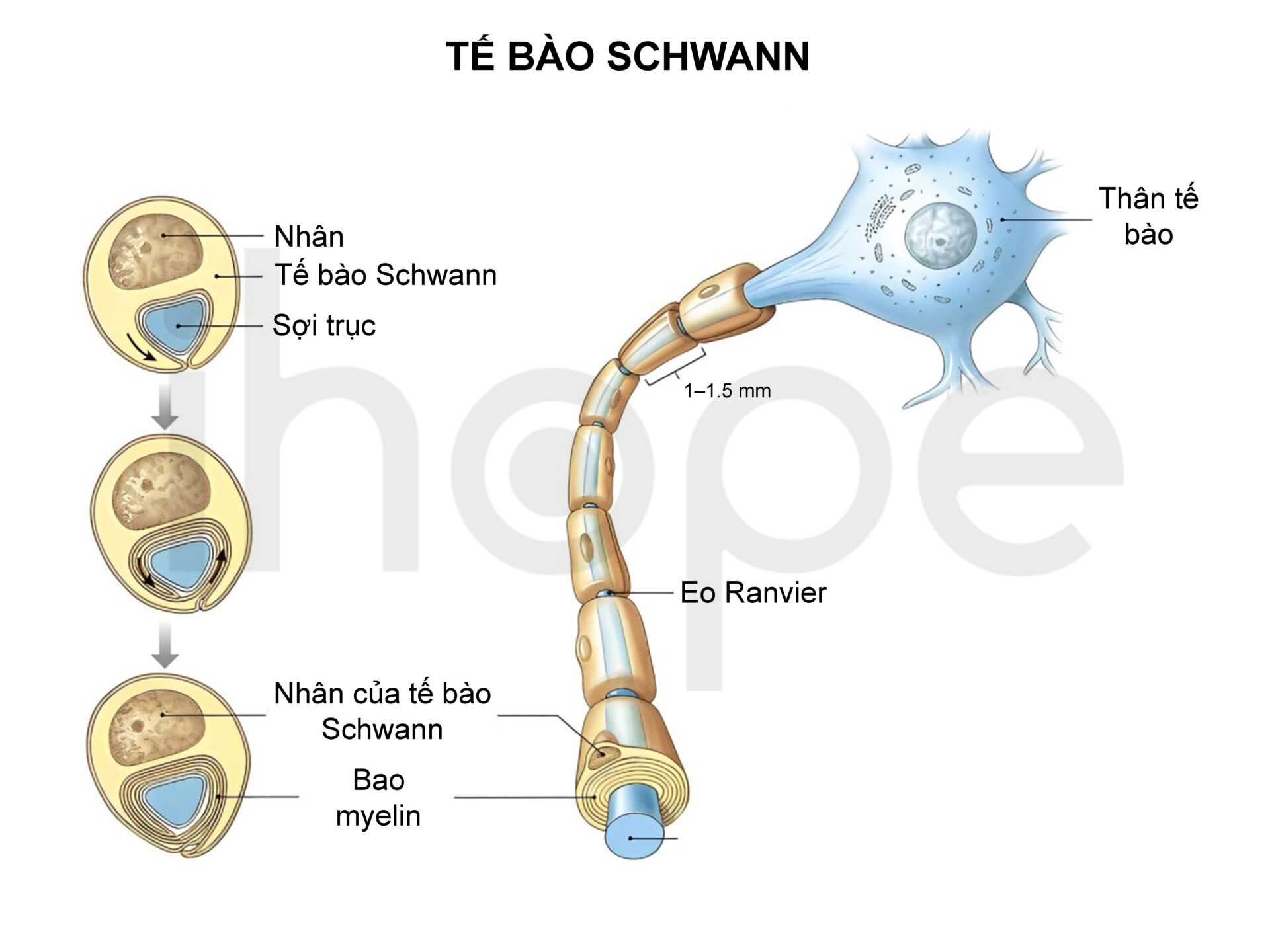
Nguồn: Simply Psychology
Phụ nữ có khả năng mắc một bệnh ung thư buồng trứng hiếm gặp là ung thư tế bào nhỏ buồng trứng loại tăng canxi máu.
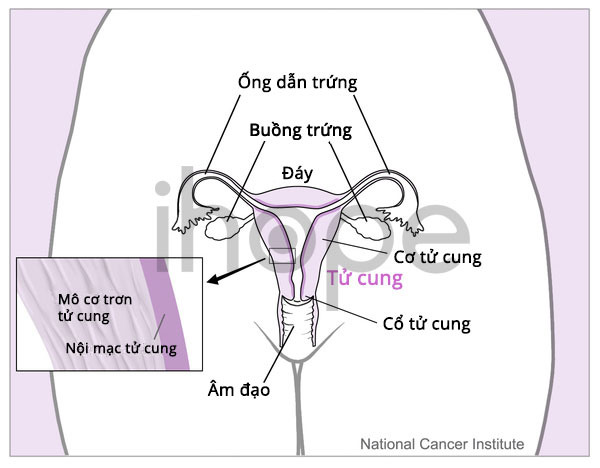
Nguồn: National Cancer Institute
Độ phổ biến
Tại Hoa Kì, ước tính tỉ lệ trẻ em dưới 15 tuổi mắc khối u cơ vân khoảng 1/1.000.000. Trong đó, hội chứng khuynh hướng khối u cơ vân chiếm từ 1/3–1/4 các trường hợp trên.
Nguyên nhân
Đột biến gen SMARCB1 gây ra phần lớn các trường hợp mắc hội chứng khuynh hướng khối u cơ vân (chiếm 85–95%). Các trường hợp còn lại do đột biến gen SMARCA4 gây ra. Hai gen này cung cấp hướng dẫn tạo ra protein có chức năng hình thành nên các tiểu đơn vị của phức hợp protein SWI/SNF. Phức hợp SWI/SNF điều phối hoạt động của gen thông qua quá trình tái cấu trúc chất nhiễm sắc.
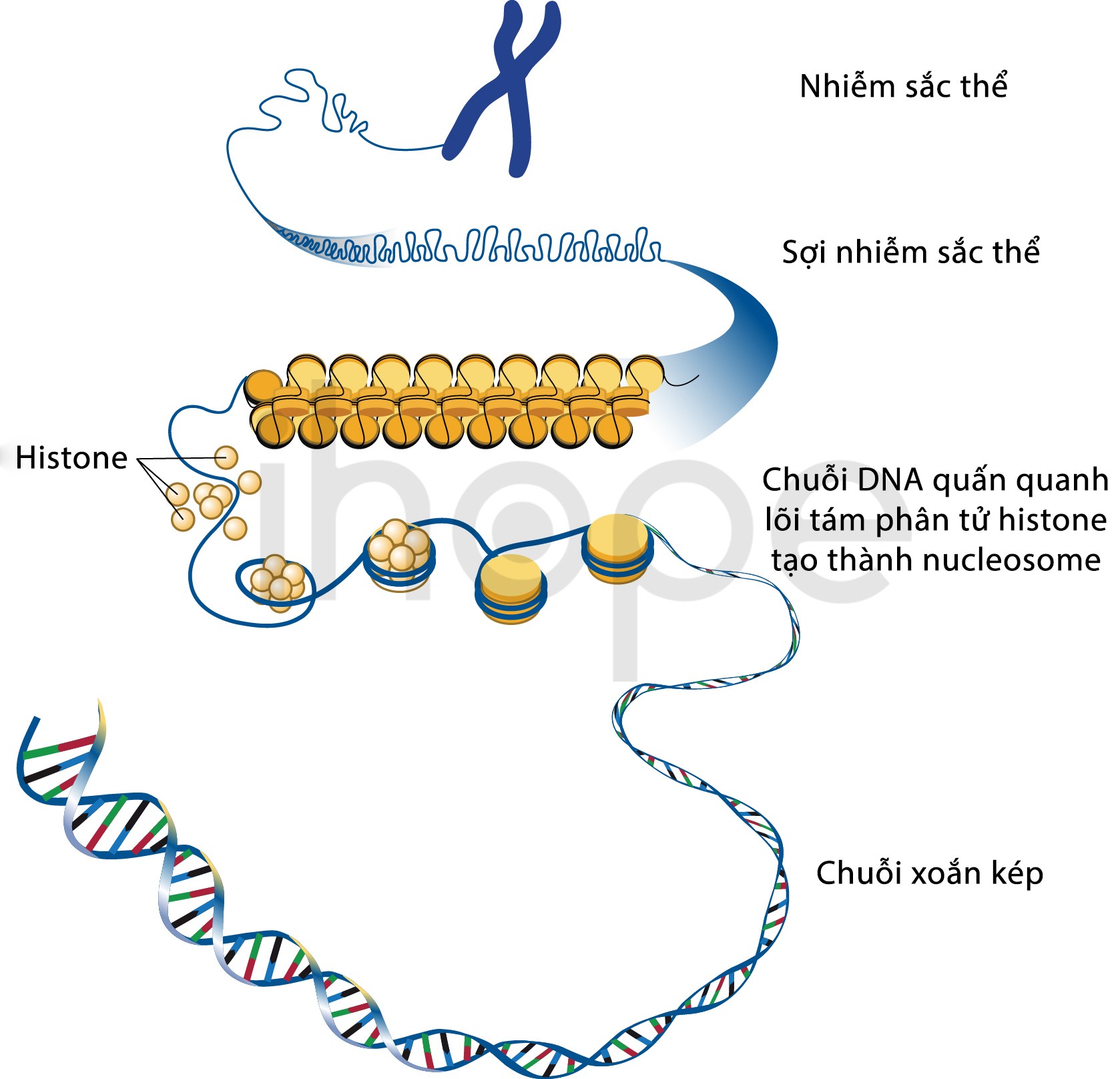
Chất nhiễm sắc nằm trong nhiễm sắc thể, chúng chứa ADN và protein. ADN mang thông tin di truyền của tế bào. Protein chính trong chất nhiễm sắc là histon, chúng giúp đóng gói ADN thành dạng nhỏ gọn phù hợp với nhân tế bào. Những thay đổi trong cấu trúc nhiễm sắc có liên quan đến quá trình sao chép ADN và biểu hiện gen
Nguồn: Darryl Leja, NHGRI
Chất nhiễm sắc là mạng lưới ADN và protein, chúng có chức năng đóng gói ADN thành nhiễm sắc thể. Chất nhiễm sắc có thể thay đổi cấu trúc nhằm kiểm soát mức độ đóng chặt của ADN. Do đó, tái cấu trúc chất nhiễm sắc là cơ chế điều hòa biểu hiện gen trong quá trình phát triển tế bào. Khi ADN đóng gói chặt, biểu hiện gen sẽ thấp hơn so với ADN đóng gói lỏng.
Dựa vào khả năng điều chỉnh hoạt động gen, phức hợp SWI/SNF tham gia nhiều hoạt động như sửa chữa ADN hư hỏng, sao chép ADN , kiểm soát quá trình tăng trưởng, phân chia và biệt hóa tế bào.
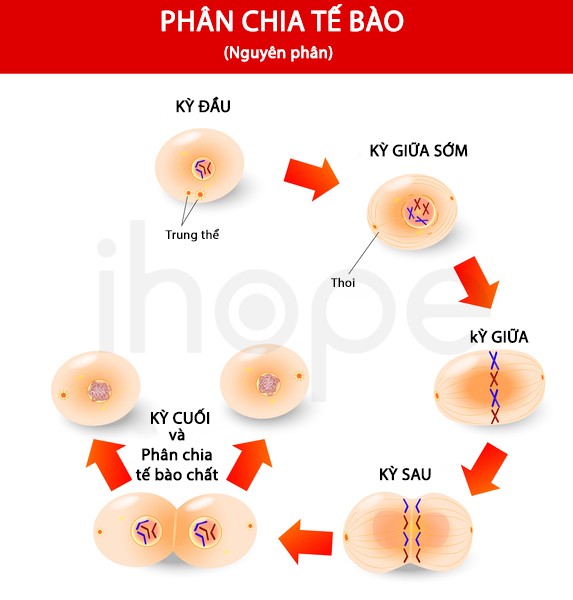
Nguồn: Designua/Shutterstock.com
Ngoài ra, gen SMARCB1 và SMARCA4 còn sản xuất các protein có chức năng như chất ức chế khối u, chúng ngăn chặn tế bào phát triển và phân chia mất kiểm soát.
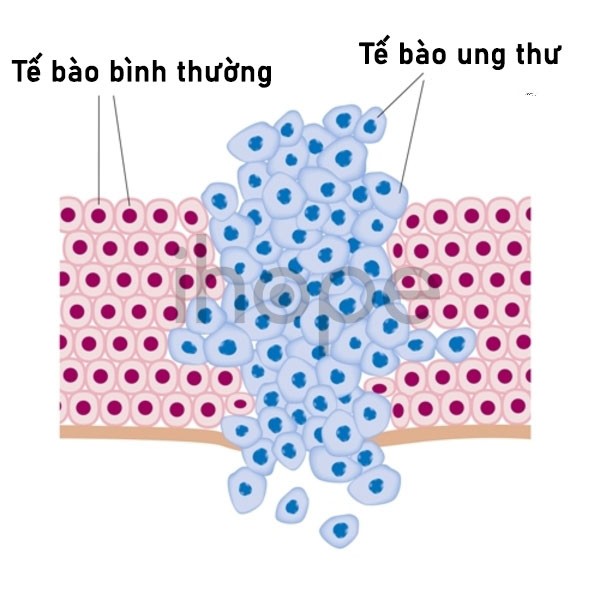
Nguồn: Alila Medical Media/Shutterstock.com
Hội chứng khuynh hướng khối u cơ vân xảy ra do đột biến gen SMARCB1 hoặc SMARCA4 theo dòng mầm, nghĩa là đột biến di truyền từ cha mẹ và chúng hiện diện trong tất cả tế bào của cơ thể. Tuy nhiên, để khối u phát triển cần thêm đột biến mất một bản sao của gen SMARCB1 hoặc SMARCA4. Đột biến này xảy ra sau khi sinh, không di truyền và chỉ hiện diện trong tế bào ung thư (đột biến soma).
Đột biến dòng mầm và đột biến soma kết hợp với nhau tạo ra protein có chức năng bất thường hoặc ngăn chặn gen SMARCB1 hoặc SMARCA4 sản xuất protein. Nếu cơ thể thiếu protein ức chế khối u, tế bào sẽ phát triển, phân chia mất kiểm soát rồi hình thành nên khối u cơ vân.
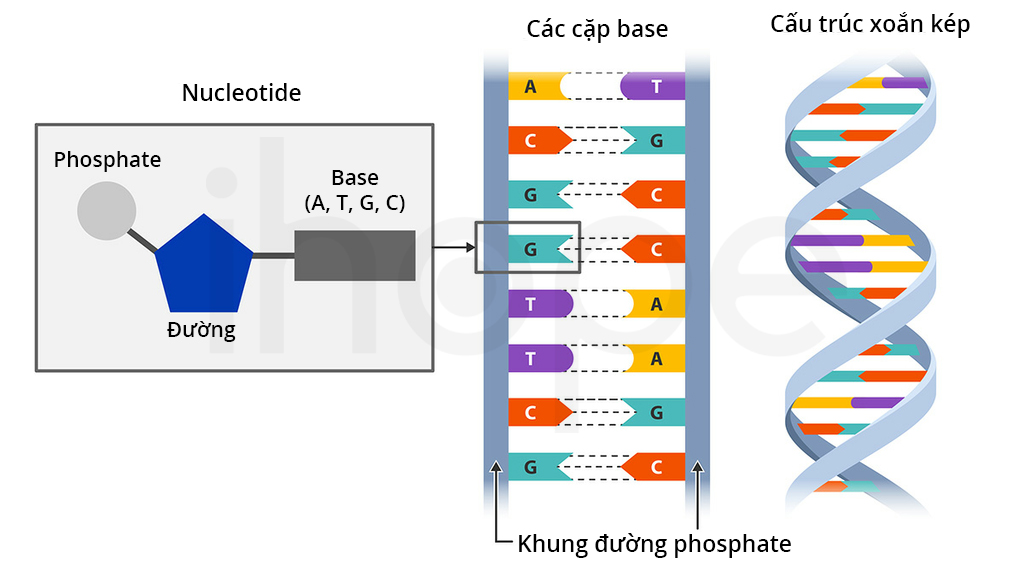
Ảnh: Cấu trúc ADN
Nguồn: Labster Theory pages
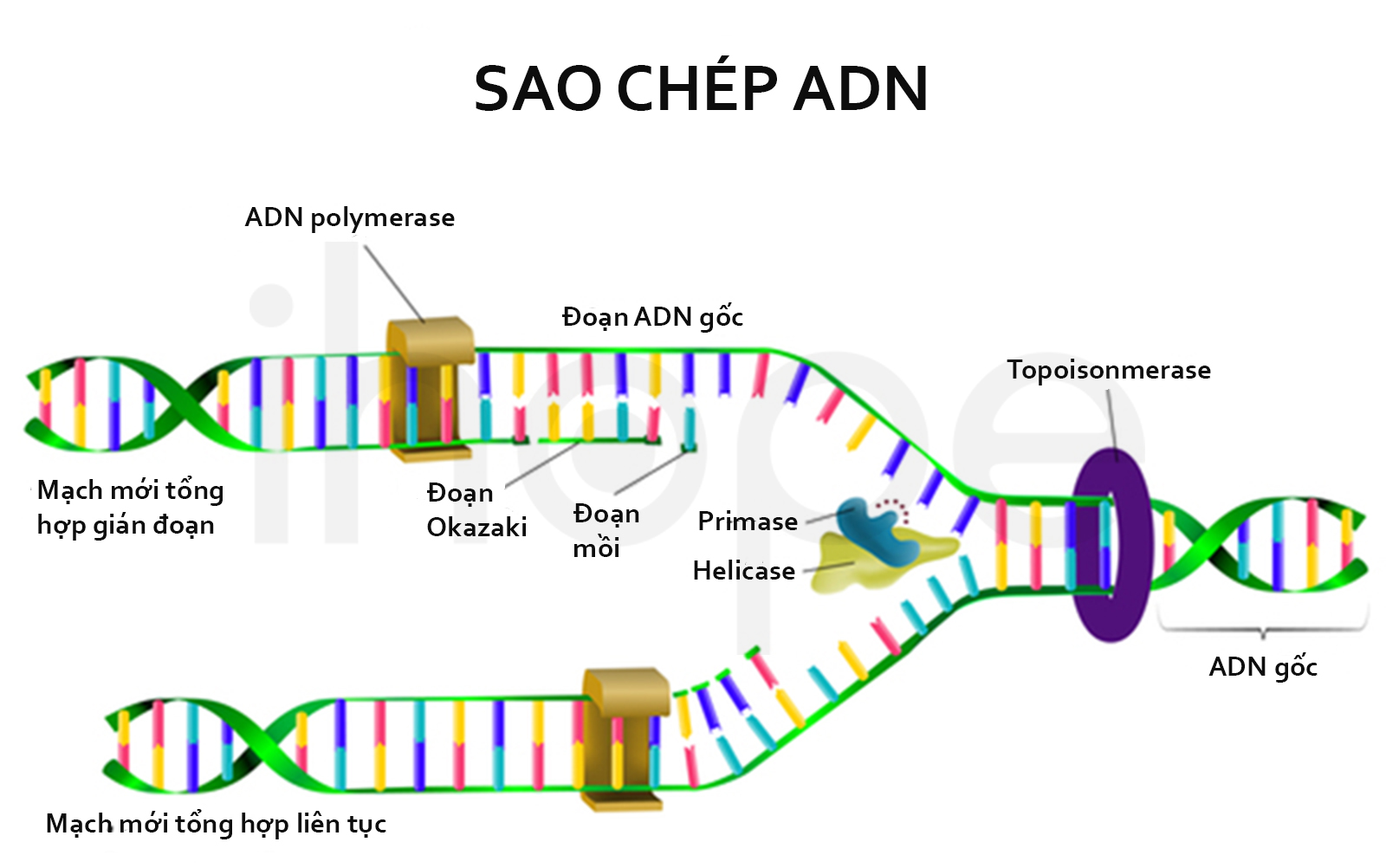
Ảnh: Quá trình sao chép ADN
Nguồn: Designua/Shutterstock.com
Chẩn đoán
Bác sĩ thường chẩn đoán hội chứng khuynh hướng khối u cơ vân dựa trên các biểu hiện lâm sàng và tiền sử bệnh gia đình.
Một người có nguy cơ cao mắc bệnh nếu có các điểm sau:
- Khối u cơ vân bẩm sinh: chẩn đoán trước sinh hoặc trong vòng 28 ngày sau sinh
- Khối u cơ vân khởi phát sớm: xuất hiện trên trẻ dưới 12 tháng tuổi
- Có khối u cơ vân tiến triển tại thời điểm chẩn đoán
- Có nhiều hơn một khối u cơ vân nguyên phát
- Tiền sử gia đình mắc các bệnh ác tính như khối u cơ vân, u bạch cầu tủy xương, u màng não
Ngoài ra, người bệnh cần thực hiện các xét nghiệm hình ảnh như siêu âm vùng bụng hoặc MRI toàn thân nhằm phát hiện khối u hiện diện trong cơ thể. Bên cạnh đó, xét nghiệm hóa mô miễn dịch trên mô khối u có thể được dùng để đánh giá mức độ thiếu protein chức năng liên quan đến hội chứng này.
Bác sĩ có thể chỉ định bệnh nhân làm xét nghiệm di truyền nhằm xác nhận kết quả chẩn đoán. Cụ thể hơn, bác sĩ thu nhận mẫu máu người bệnh để tách chiết ADN, sau đó, hai bản sao của gen SMARCB1 được phân tích nhằm tìm kiếm các đột biến liên quan. Trường hợp ADN được tách chiết trực tiếp từ mô khối u, hai bản sao của gen SMARCB1 được khảo sát để tìm kiếm các đột biến liên quan đến quá trình hình thành khối u.
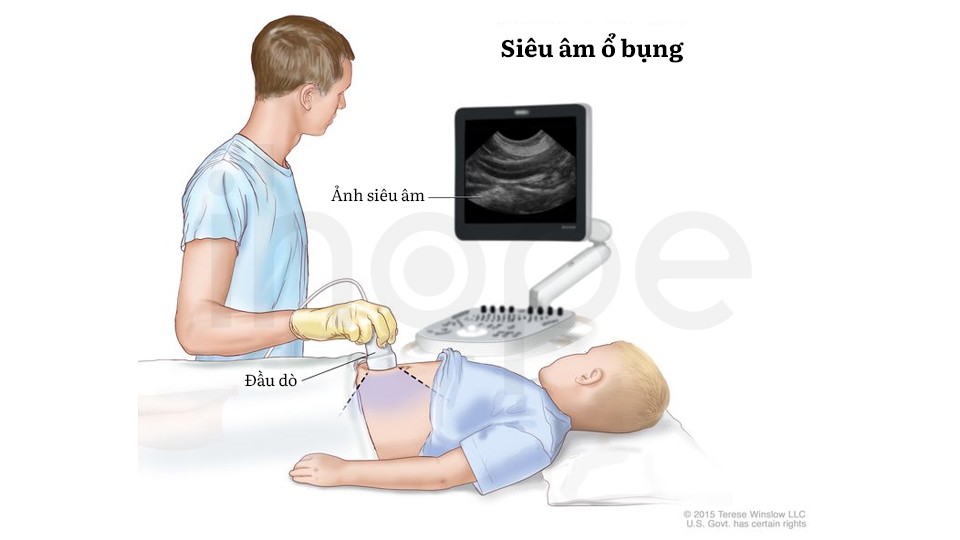
Ảnh: Siêu âm ổ bụng
Nguồn: © 2015 Terese Winslow LLC for the National Cancer Institute
Điều trị
Điều trị hội chứng khuynh hướng khối u cơ vân cần kết hợp nhiều phương pháp với nhau. Các liệu pháp điều trị phổ biến bao gồm:
Dạng di truyền
Hội chứng khuynh hướng khối u cơ vân di truyền theo kiểu trội trên nhiễm sắc thể thường. Do đó, đột biến trên một bản sao của gen trong mỗi tế bào đủ để gây ra bệnh. Đa phần các trường hợp mắc bệnh do đột biến gen SMARCA4, người bệnh được di truyền từ cha hoặc mẹ mắc bệnh. Tuy nhiên, cha hoặc mẹ mang gen đột biến này thường không biểu hiện triệu chứng hay phát triển khối u cơ vân.

Nguồn: U.S. National Library of Medicine
Đối với trường hợp người bệnh mang đột biến gen SMARCB1, bệnh do đột biến mới (denovo) xuất hiện trong quá trình hình thành tế bào sinh sản (trứng, tinh trùng) hoặc quá trình phát triển phôi sớm.
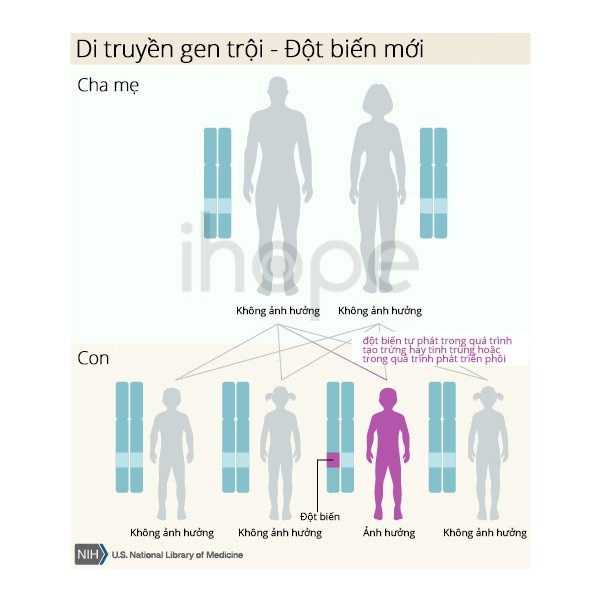
Nguồn: U.S. National Library of Medicine
Phòng ngừa
Hội chứng khuynh hướng khối u cơ vân di truyền trội trên nhiễm sắc thể thường, nếu cha hoặc mẹ mắc bệnh, con sinh ra sẽ có 50% khả năng di truyền bệnh. Do đó, để bảo đảm 100% khả năng con không bị bệnh, cha mẹ có thể chọn phương pháp thụ tinh nhân tạo IVF và sàng lọc phôi PGS/PGD. Các thành viên trong gia đình nên khám sức khỏe và tầm soát bệnh định kì nếu có thành viên mắc bệnh. Các cặp vợ chồng trước khi mang thai cần tư vấn và xét nghiệm di truyền nhằm đảm bảo sinh con khỏe mạnh.
Ngoài ra, người bệnh nên duy trì lối sống lành mạnh, kiểm tra định kì và tầm soát ung thư nhằm phát hiện bệnh sớm và điều trị kịp thời.
Một số biện pháp nhằm hạn chế nguy cơ mắc bệnh bao gồm:
- Chế độ ăn uống lành mạnh với nhiều trái cây và rau quả
- Tập thể dục thường xuyên
- Không hút thuốc
- Tránh tiếp xúc với khói thuốc
- Chú ý bôi kem chống nắng, đội mũ và mặc quần áo bảo hộ khi ra ngoài
Ngoài ra, người có nguy cơ cao mắc bệnh nên theo dõi sức khỏe bản thân nhằm phát hiện sớm các dấu hiệu cảnh báo bệnh như:
- Chán ăn
- Sụt cân không rõ nguyên nhân
- Đau nhức, nổi cục hoặc sưng tấy không rõ nguyên nhân
- Nhức đầu, thay đổi thị lực hoặc rối loạn chức năng thần kinh
Các tên gọi khác
- RTPS
- Familial rhabdoid tumor
- Rhabdoid predisposition syndrome
- Hereditary SWI/SNF deficiency syndrome
- Familial posterior fossa brain tumor syndrome
- Familial posterior fossa brain tumor of infancy
References
- Genetic Testing Information. Rhabdoid tumor predisposition syndrome 1. Retrieved August 12, 2024 from https://www.ncbi.nlm.nih.gov/gtr/conditions/C1836327/
- Genetic and Rare Diseases Information Center. Rhabdoid tumor. Retrieved August 12, 2024 from https://rarediseases.info.nih.gov/diseases/7572/index
- Catalog of Genes and Diseases from OMIM. RHABDOID TUMOR PREDISPOSITION SYNDROME 1; RTPS1. Retrieved August 12, 2024 from https://omim.org/entry/609322
- U.S National Library of Medicine. Rhabdoid tumor predisposition syndrome. Retrieved August 12, 2024 from https://medlineplus.gov/genetics/condition/rhabdoid-tumor-predisposition-syndrome/
- Children's Hospital of Philadelphia. Rhabdoid Tumor Predisposition Syndrome. Retrieved August 12, 2024 from https://www.chop.edu/conditions-diseases/rhabdoid-tumor-predisposition-syndrome
- Frontiers. Rhabdoid Tumor Predisposition Syndrome: From Clinical Suspicion to General Management. Retrieved August 12, 2024 from https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2021.586288/full
- National Cancer Institute. Rhabdoid Tumor Predisposition Syndrome Type 1 (PDQ®)–Health Professional Version. Retrieved August 12, 2024 from https://www.cancer.gov/publications/pdq/information-summaries/genetics/rtps1-hp-pdq
- National Institute of Health. Rhabdoid Tumor Predisposition Syndrome. Retrieved August 12, 2024 from https://www.ncbi.nlm.nih.gov/books/NBK469816/